










 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
Un pseudolinfoma es una proliferación linfocítica benigna, reversible, inflamatoria, reactiva y policlonal que regresa espontáneamente o cura tras la eliminación del factor causal.1 Los pseudolinfomas de la piel son proliferaciones linfocíticas benignas de células B o T que simulan linfomas cutáneos clínica e histológicamente. Se han utilizado numerosos sinónimos para la designación de pseudolinfomas de la piel: sarcoide de Spiegler-Fendt, linfocitoma cutis, linfadenosis benigna cutánea e hiperplasia linfoide cutánea.2
Las lesiones varían en cuanto a número, características clínicas, aspecto histológico y posible etiología. Clínicamente, pueden observarse pápulas, placas y tumores de distribución solitaria, regional o diseminada. Algunos pseudolinfomas simulan lesiones eccematosas o linfomas de células T cutáneos en etapa temprana. El cuadro histológico puede simular el patrón clásico de los linfomas cutáneos de células B o de células T.2,3
Con frecuencia es de causa idiopática, pero los principales factores causales asociados son picaduras de insectos, borreliosis, traumas, vacunas, reacción a drogas, tatuajes, aretes de oro, acupuntura y cicatrices de infección por el virus de la varicella-zóster. Es más frecuente en la cara (mejillas, nariz, lóbulo de la oreja), pecho y extremidades superiores y predomina en mujeres (3:1) y en personas de piel blanca (9:1).4
Los criterios histológicos para el diagnóstico de pseudolinfoma cutáneo incluyen dos rasgos principales: el patrón arquitectural del infiltrado y su composición celular, que con frecuencia muestra un carácter mixto. La diferenciación clara entre los infiltrados linfoides benignos y malignos de la piel solo es posible después de la síntesis y la integración de las características clínicas, histopatológicas, immunofenotípicas y moleculares. En algunos casos, solo el seguimiento cuidadoso del paciente y la respuesta al tratamiento permiten establecer el diagnóstico de certeza.1,5
La plasmocitosis cutánea representa una variante peculiar e infrecuente de pseudolinfoma. Se ha descrito con mayor frecuencia en japoneses, es muy rara en caucásicos, consta de una triada de lesiones cutáneas, linfadenopatía superficial e hiperganmaglobulinemia policlonal, predomina en hombres, el rango de edades es de 20 a 68 años y raramente se presenta en niños. Clínicamente se manifiesta como lesiones cutáneas de color marrón rojizo, pruriginosas, en forma de máculas, pápulas, placas o nódulos, persistentes, en el tronco y las extremidades superiores, o, con menor frecuencia, diseminadas;4 se informan casos aislados con presentación en forma de nódulos ulcerados.6
Histológicamente se caracteriza por un infiltrado dérmico linfoplasmacítico con disposición perivascular o nodular que puede extenderse al subcutis, con numerosas células plasmáticas maduras policlonales. Los pacientes afectados pueden tener asociados hallazgos sistémicos (fiebre, adenopatías e hipergammaglobulinemia). Algunos pacientes desarrollan plasmocitosis sistémica, caracterizada por afectación multiorgánica por infiltrados ricos en células plasmáticas policlonales. Por definición, no existe atipia citológica ni actividad mitótica. Pueden estar presentes linfocitos e histiocitos en pequeño número y se observan folículos linfoides en algunos casos. Con técnicas de immunohistoquímica las células plasmáticas son policlonales y la tinción para herpes virus humano 8 (HHV8) es negativa. La médula ósea y los ganglios linfáticos muestran infiltración por células plasmáticas maduras policlonales y no atípicas.4
Generalmente tiene un pronóstico favorable, aunque algunos casos tienen un curso clínico más agresivo, con infiltración visceral. Cuando la plasmocitosis primaria afecta más de dos órganos se denomina plasmocitosis sistémica (se ha informado afectación pulmonar y de ganglios linfáticos). Se ha descrito su asociación con neumonía intersticial, tuberculosis y sífilis.7 La eritrosedimentación está elevada usualmente y en algunos pacientes se detectan anticuerpos antinucleares. Los exámenes para excluir un mieloma son invariablemente negativos.4,7,8 No se ha informado la transformación de la plasmocitosis cutánea en linfoma maligno o en mieloma múltiple.8
En un estudio realizado en Japón la población de células plasmáticas en la médula ósea estuvo entre 1,6 y 12%. Este parámetro se considera un predictor de la evolución de la enfermedad, junto a la concentración sérica de inmunoglobulinas.9 No existe un método uniforme para el tratamiento de la plasmocitosis. Se han utilizado esteroides sistémicos y tópicos, radiciones, tacrolimus, antibióticos, quimiotrapia sistémica y terapia con anticuerpos anti CD20. Recientemente se describe el uso de tocilizumab (antagonista de interleucina -6) con mejoría sintomática de las lesiones.10
INFORMACIÓN DEL PACIENTE
Paciente de 80 años, femenina, con antecedentes de polimialgia reumática e hipertensión arterial, que acudió a la Consulta de Dermatología del Hospital Provincial Universitario Clínico Quirúrgico “Arnaldo Milián Castro” de la Ciudad de Santa Clara, Provincia de Villa Clara, por presentar lesiones cutáneas con prurito marcado y ardor, asociadas a edema periorbitario, de ocho meses de evolución. Llevaba tratamiento con esteroides sistémicos, antihistamínicos y cremas esteroideas, pero mostraba una evolución crónica, con persistencia de las lesiones.
Hallazgos clínicos
Al examen físico se constataron lesiones cutáneas eritematosas y habonosas, diseminadas en el tórax, el abdomen y los miembros. No presentaba adenopatías ni hepatoesplenomegalia.
Evaluación diagnóstica
Los exámenes de laboratorio mostraron anemia (hemoglobina: 7,7 g/l) y un leucograma normal. El medulograma descartó el diagnóstico de mieloma de células plasmáticas (proporción de plasmocitos del 3%).
El examen histológico de una de las lesiones cutáneas en la piel del escote mostró un infiltrado celular dermal, perivascular e intersticial, con patrón nodular compuesto principalmente por células plasmáticas maduras con aislados linfocitos e histiocitos. (Figura 1 y Figura 2). El estudio inmunohistoquímico reveló la policlonalidad de las células plasmáticas al presentar tinción citoplasmática para ambas cadenas ligeras de inmunoglobulinas (kappa y lambda) -Figura 3-.
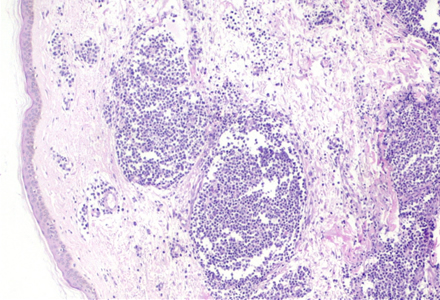
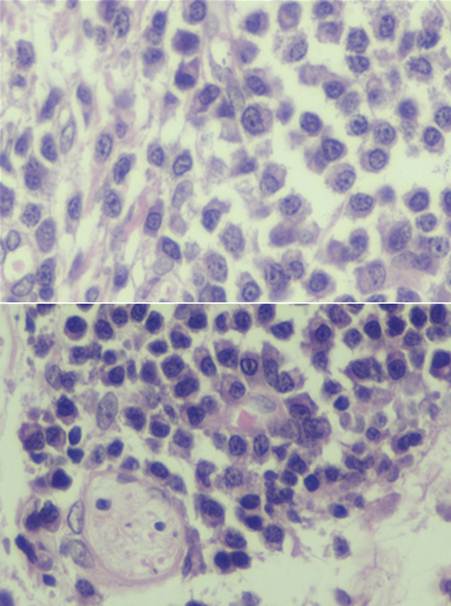
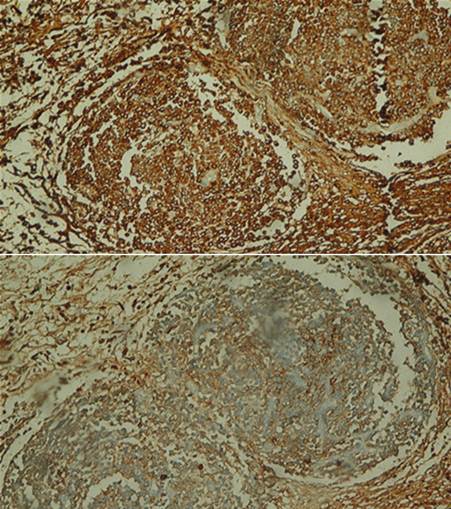
Intervención terapéutica
Llevó tratamiento con esteroides orales, antihistamínicos y cremas esteroideas.
Seguimiento y resultados
La paciente mantiene seguimiento por Hematología y Dermatología. Mostró mejoría clínica de las lesiones, pero no se logró una completa remisión.
DISCUSIÓN
En la literatura solo se han documentado menos de un centenar de casos de plasmocitosis cutánea y sistémica.10 En esta paciente la presentación clínica de las lesiones fue similar a la descrita por otros autores,5,8,9,10 con la diferencia de que existen, además, lesiones en diana que simularon un eritema multiforme. No se indicaron estudios para determinar los valores de inmunoglobulinas plasmáticas, por lo que no se pudo comprobar la presencia de hiperganmaglobulinemia.
La plasmocitosis cutánea debe diferenciarse del mieloma de células plasmáticas, el plasmocitoma cutáneo y el linfoma de zona marginal cutáneo, los que pueden mostrar un número significativo de células plasmáticas, pero son monoclonales y con frecuencia atípicas. La variante de células plasmáticas de la enfermedad de Castleman multicéntrica con afectación de la piel muestra características comunes con la plasmacitosis cutánea; no obstante, la enfermedad de Castleman es una enfermedad más severa, con anemia, trombocitopenia y asociación con la infección por el virus de inmunodeficiencia humana y el del herpes humano 8.4 Esta enfermedad también se debe diferenciar de los infiltrados dérmicos de células plasmáticas asociados a infecciones como sífilis y enfermedad de Lyme y de las enfermedades del tejido conectivo. Los infiltrados en estos trastornos tienden a ser más esparcidos y la correlación clínica es usualmente discriminatoria.4

