










Meu SciELO
 Serviços customizados
Serviços customizadosServiços Personalizados
Artigo
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkRevista Cubana de Medicina
versão On-line ISSN 1561-302X
Rev cubana med v.42 n.4 Ciudad de la Habana jul.-ago. 2003

Hospital Clinicoquirúrgico "Hermanos Ameijeiras"
Síndrome de linfohistiocitosis hemofagocítica: a propósito de un caso
Dra. Zulema González del Valle González,1 Dr. Alfredo Vázquez Vigoa,2 Dr. Rolando Rodríguez Fernández,3 Dr. Luis Javier Lima Pérez4 y Dr. Isidro Machado Puerto5
Resumen
El síndrome de linfohistiocitosis hemofagocítica (LHH), aunque infrecuente en la práctica clínica, resulta de extraordinario interés dado su comportamiento agresivo y mal pronóstico. El objetivo de esta publicación fue exponer, a partir de la experiencia derivada de la observación de un caso, las características clínicas fundamentales: anemia, trombocitopenia y neutropenia unido a fiebre y esplenomegalia. Se discutieron las causas que pueden producir dicho síndrome y entre ellas los linfomas, como sucedió en este caso. Se concluyó que el síndrome de LHH constituye una entidad en específico y que se necesita una búsqueda minuciosa desde el punto de vista etiológico para determinar su causa. Se destacó que este caso fue secundario a un linfoma no Hodking extranodal de células T de alto grado de malignidad con toma renal que llevó a la muerte a la enferma en breve período.
DeCS: HISTIOCITOSIS DE CELULAS NO LANGERHANS/etiología; HISTIOCITOSIS DE CELULAS NO LANGERHANS/mortalidad; LINFOMA NO HODGKING; ANEMIA; NEUTROPENIA; TROMBOCITOPENIA; ESPLENOMEGALIA.
El síndrome de linfohistiocitosis hemofagocítica (LHH), denominado también reticulohistiocitosis histiocítica medular, fue descrito por primera vez en 1939;1,2 se referían a un agrupamiento inusual de manifestaciones clínicas caracterizadas por fiebre, esplenomegalia, artralgias, citopenia y hallazgo patológico de hemofagocitosis expresada por macrófagos que fagocitan eritocitos, leucocitos y plaquetas. El síndrome de LHH puede aparecer de manera primaria,3 aunque es más frecuente la forma secundaria, se señalan las infecciones como causa primordial.4,5 A continuación resumimos las características fundamentales de un caso.
Presentación del caso
Paciente de raza blanca, sexo femenino, 56 años de edad, que ingresa por fiebre, decaimiento marcado y palidez cutáneo mucosa.
Historia de la enfermedad actual
Paciente con antecedentes de buena salud que se encontraba trabajando en Sudáfrica, y que 30 d previos a su ingreso presentó un cuadro diarreico con escalofríos. Dos semanas después comenzó a presentar fiebre de 38,5-39 °C, fundamentalmente por las noches, acompañada de molestias abdominales, astenia, anorexia y lesiones cutáneas distribuidas por el tronco y las extremidades. Se le practicaron algunos exámenes, incluido un medulograma, no se concluyó el diagnóstico. Se le trasladó a nuestro país y se decidió su ingreso en este centro.
Examen físico
Palidez cutáneo mucosa marcada con discreto tinte subictérico de escleróticas, hepatosplenomegalia importante a predominio esplénico y edemas en miembros inferiores de fácil godet.
Exámenes complementarios
Hemograma: hto. 0,24; leucocitos 2,3 x 109/L; P 0,69; L0,27; M0,04 y E0,00. Se observan células de aspecto blástico.
Glucosa: 5,0mmol/L, creatinina: 93mmol/ L.
TGO: 26 U/ L; TGP: 10,3 U/ L; FAL: 130 U/ L; LDH :728U/L.
Proteínas totales: 62 g/ L; albúmina: 32 g/ L.
TP: 16 s. TPK: 43 s.
Fibrinógeno: 1,57 g/ L.
Serología para VIH: negativa.
Serología para EBV: negativa.
Gota gruesa para paludismo: negativa.
Serología para CMV: negativa.
Hemocultivos I, II, III: negativos.
Medulograma: anisopoiquilocitosis, hipocromía, trombopenia severa. Sistema megacariopoyético hiperplástico; granulopoyético: íntegro; eritropoyético: hiperlasia severa. Azul de prusia: positivo. Celularidad: hipercelular, se observa aumento de células plasmáticas y reticulohistiocitarias. Se concluye como médula con elementos de reactividad, llama la atención el aumento de los histiocitos y el fenómeno de hemofagocitosis. No infiltración tumoral.
Biopsia de médula ósea: médula hipercelular global con hiperplasia de las 3 series de aspecto reactivo. Aumento de los histiocitos de forma difusa y fenómeno de hemofagocitosis.
Cituria: negativa.
Addis 2 h: L 333 H2500 C 0 Prot 0,55mg/min.
Ultrasonido de hemiabdomen superior HAS: hepatosplenomegalia importante con cambios de la ecogenicidad hepática, vesícula biliar y resto de las vías biliares sin alteraciones. No se detectan adenopatías intra-abdominales, no hay líquido libre en cavidad abdominal.
Ecocardiograma: calcificación de la valva y anillo posterior de la mitral, no dilatación de cavidades, no hipertrofia del ventrículo izquierdo, no derrame pericárdico.
Evolución en sala
La paciente se mantuvo muy asténica y febril, a los 7 d de su ingreso comenzó a presentar disnea, palpitaciones y persistía la fiebre con escalofríos, a pesar del tratamiento antimicrobiano, la adinamia y la disnea se exacerbaron. Comenzó a instalarse una oliguria progresiva con elevación de las cifras de creatinina por encima de 300 mmol/L, lo que motivó su traslado a la unidad de cuidados intermedios, donde requirió tratamiento dialítico. Se instaló fallo respiratorio y falleció.
Discusión
En la actualidad se plantea la necesidad de reunir criterios para el diagnóstico del síndrome de LHH, los cuales incluyen elementos clínicos, de laboratorio e histopatológicos.6 La fiebre y la esplenomegalia constituyen los signos clínicos más prominentes, pero la hepatomegalia, linfadenopatía, artralgia y rash constituyen otras manifestaciones del síndrome. El rash puede ser maculopapular o en forma de erupción nodular, en ocasiones, las manifestaciones clínicas sugieren una infección viral aguda como el EBV, CMV, hepatitis viral, infección por el VIH, así como leishmaniasis, brucelosis, rickettsiosis y malaria. Las anormalidades de laboratorio incluyen citopenias, elevación de la LDH, hipertrigliceridemia, descenso del fibrinógeno y cuadro de coagulación intravascular diseminada.
Histológicamente, la hemofagocitosis puede observarse en médula ósea como se detectó en nuestro caso, así como en los tejidos.
El síndrome de LHH puede presentarse en todas las edades, aunque la forma primaria es mucho más frecuente en niños. El mecanismo patogénico, ya sea de forma primaria o secundaria, por el cual (virus, hongos, linfomas) desencadenan el fenómeno de hemofagocitosis no está bien aclarado, pero se supone que sea mediante una hiperactivación de macrófagos, los cuales comienzan a englobar células sanguíneas, a partir de una inapropiada respuesta de los TH1 a patógenos intracelulares.
A continuación exponemos los criterios clínico-patológicos para el diagnóstico del síndrome:5
- Fiebre ³ 38,5 °C por 7 o más días.
- Esplenomegalia de 3 cm o más por debajo del reborde costal
Dos de las siguientes alteraciones hematológicas:
- Anemia: menos de 9,0 g/L.
- Trombopenia: menor de 100 000 plaquetas.
- Neutropenia: menor de 10 neutrófilos.
- Hipertrigliceridemia: mayor de 2,0 mmol/ L.
- Hipofibrinogenemia menor de 1,5.
- Hemofagocitosis en médula ósea, bazo o nódulo linfático.
- No evidencia de médula ósea hipoplástica o neoplasia maligna.
Esta paciente reunió la mayoría de los criterios necesarios para el diagnóstico del síndrome, se destacó la fiebre persistente, la gran esplenomegalia, el rash maculopapular, la citopenia, la elevación de la LDH, la hipofibrinogenemia y la demostración en médula ósea del fenómeno de hemofagocitosis. El curso tormentoso de la enferma con evolución a la insuficiencia renal está determinado por la causa del síndrome (fig. 1) que resultó ser un linfoma no Hodking (fig. 3) extranodal de células T de alto grado de malignidad con invasión renal, (fig. 2) que explica el fallo renal terminal, hallazgos que fueron comprobados en el estudio necrópsico.

Fig. 1 Corte histológico de hígado con H/E a 100 x que muestra célula de Kuffer con fagocitosis de hematíes(Síndrome Hemofagocítico).
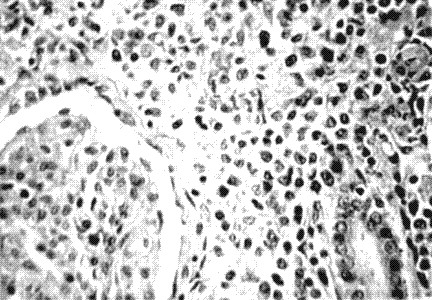
Fig. 2. Corte histológico de riñón con H/E a 40 x que muestra infiltración intersticial difusa por células tumorales linfoides que invaden los túbulos renales.
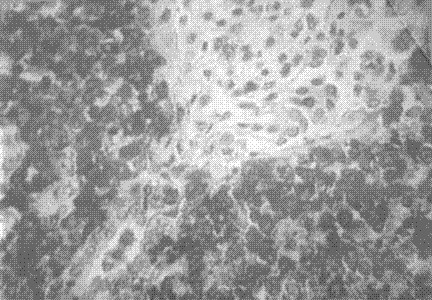
Fig. 3 Técnica de inmunohistoquímica para células T positiva en células tumorales linfoides.
Summary
The hemophagocytic lymphohistiocytosis syndrome (HLH), though unfrequent in clinical practice, is of extraordinary interest due to its aggressive behavior and poor prognosis. The objective of this paper was to expose, starting from the experience derived from the observation of a case, the fundamental clinical characteristics: anemia, thrombocytopenia and neutropenia together with fever and splenomegaly. The causes that may produce such syndromes and lymphomas, as it occurred in this case, were discussed. It was concluded that the HLH is an specific entity and that a thorough search from the etiological point of view is needed to determine its cause. It was stressed that this case was secondary to a highly malignant non-Hodgking extranodal T cell lymphoma with renal taking leading to the death of the female patient in a short period of time.
Subject headings: HISTIOCYTOSIS, NON-LANGERHANS-CELL/etiology; HISTIOCYTOSIS, NON-LANGERHANS-CELL/mortality; LYMPHOMA, NON HODGKIN; ANEMIA; NEUTROPENIA; THROMBOCYTOPENIA; SPLENOMEGALY.
Referencias bibliográficas
- Flavara BE. Hemophagocytic lymphohistiocytosis syndrome. Semin Pathol Diagn 1992;9:63-74.
- Reiner AP, Spivak JL. Hematophagic histiocytosis. A report of 23 new patients and a eview of the literature. Medicine 1988;67:369.
- Loy TS, Díaz Anas AA, Perry MC. Familial erytrophagocytic-lymphohistiocytosis. Semin Oncol 1991;18:34-9.
- Kawa K. Epstein Barr virus associated diseases in humans. Internal J Hematol 2000; 71:108-17.
- Kawaguchi H, Miyashita T, Herbs H. Epstein Barr virus infected T lymphocytes in Epstein Barr virus associated hemophagocytic syndrome. J Clin invest 1993; 92:1444-50.
- Henter JI, Elinder G, and the FHL study group of the Histiocyte Society. Diagnostic Guidelines for hemophagocytic lympho-histiocytosis. Sem Oncol 1991;18:34-9.
Recibido: 13 de septiembre de 2002. Aprobado: 7 de enero de 2003.
Dra. Zulema González del Valle González. Hospital Clinicoquirúrgico "Hermanos Ameijeiras" San Lázaro No. 701 entre Belascoaín y Marqués González, Centro Habana, Ciudad de La Habana, Cuba.
1 Especialista de I Grado en Medicina Interna. Máster en Infectología.
2 Especialista de II Grado en Medicina Interna. Profesor Auxiliar.
3 Especialista de I Grado en Medicina Interna.
4 Especialista en Medicina General Integral. Residente de 4to. Año de Medicina Interna.
5 Residente de 4to. Año de Anatomía Patológica.

