










Meu SciELO
 Serviços customizados
Serviços customizadosServiços Personalizados
Journal

Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkRevista de Ciencias Médicas de Pinar del Río
versão On-line ISSN 1561-3194
Rev Ciencias Médicas vol.17 no.3 Pinar del Río maio-jun. 2013
ARTÍCULO ORIGINAL
Caracterización del síndrome de Ehlers-Danlos tipo III
Characterization of Type-III Ehlers–Danlos Syndrome
Mirta Caridad Campo Díaz1, Abel Fortún Campo2, Aimara Beades Martínez3, Yumnary Gato Santiesteban4, César Valdés Sojo5
1Especialista de Segundo Grado en Hematología y Pediatría. Investigadora Auxiliar. Máster en Atención Integral al Niño. Profesora Auxiliar y Consultante. Hospital Provincial Pediátrico “Pepe Portilla”. Pinar del Río. Correo electrónico: mcampo@princesa.pri.sld.cu
2Especialista de Primer Grado en Medicina General Integral y Oftalmología. Máster en Longevidad Satisfactoria. Instructor. Hospital General Docente “Abel Santamaría”. Pinar del Río. Correo electrónico: adalberto76@princesa.pri.sld.cu
3Especialista de Primer Grado en Medicina General Integral y Oftalmología. Máster en Longevidad Satisfactoria. Hospital General Docente “Abel Santamaría”. Pinar del Río. Correo electrónico: abm@princesa.pri.sld.cu
4Especialista de Primer Grado en Medicina General Integral y Hematología. Instructora. Hospital Provincial Pediátrico “Pepe Portilla”. Pinar del Río. Correo electrónico: yumnag@infomed.sld.cu
5Especialista de Primer Grado en Hematología. Hospital Pediátrico Docente “Pepe Portilla”. Pinar del Río.
Correo electrónico: cvsojo@princesa.pri.sld.cu
RESUMEN
Introducción: el síndrome de Ehlers-Danlos es un grupo heterogéneo de trastornos hereditarios del tejido conectivo causado por mutaciones en los genes que codifican el colágeno fibrilar o las enzimas comprometidas en la modificación post-translacional de dichos colágenos caracterizado por afectación de la piel, las articulaciones y los vasos sanguíneos. Se manifiesta frecuentemente por un cuadro clínico de hipermovilidad articular con hiperextensibilidad asociado a anomalías cutáneas.
Objetivo: evaluar si existe un defecto de la coagulación específico que forme parte de la enfermedad para definir un diagnóstico precoz, seguimiento y tratamiento en las distintas instancias de salud.
Material y método: con el objetivo de caracterizar el síndrome en nuestra provincia se realizó una investigación aplicada, observacional, descriptiva y transversal de 305 niños con síndrome de Ehlers-Danlos tipo III en edades comprendidas entre 5 y 18 años para el diagnóstico de las manifestaciones articulares típicas de la enfermedad así como evaluación por cardiología, oftalmología, fisiatría y ortopedia realizándose además coagulograma y lámina periférica.
Resultados: no se encontró predominio de género y los antecedentes familiares sugestivos de la enfermedad estuvieron presentes en casi la mitad de los pacientes, predominando la vía materna. La historia de tendencia familiar hemorrágica fue positiva en 36 casos. Las manifestaciones encontradas en los pacientes fueron las descritas para la entidad, destacándose la existencia de sangramiento predominantemente cutáneo-mucoso o por procederes invasivos en 181 niños asociado a la presencia de macroplaquetas y plaquetas dispersas en el extendido periférico.
Conclusiones: los resultados obtenidos indican la existencia de trastornos cualitativos plaquetarios que debe evaluarse mediante estudios más específicos de la coagulación no disponibles en la provincia que incluya la agregación y la función plaquetaria.
DeCS: Síndrome de Ehlers-Danlos Tipo III; Trastornos de la coagulación.
ABSTRACT
Introduction: Ehlers–Danlos Syndrome (EDS) is a heterogeneous group of inherited connective tissue disorders, caused by mutations in the genes that codify fibrillar collagen or the enzymes implicated in modifying post-translational collagen that affects skin, joints and blood vessels. Frequently, the clinical picture presents articular hypermobility with hyperextension associated with cutaneous anomalies.
Objective: to characterize Type-III Ehlers–Danlos Syndrome in Pinar del Rio province.
Material and method: an applied, observational, descriptive and cross-sectional study that included 305 children (from 5 to 18 years old) suffering from type-III Ehlers–Danlos Syndrome (EDS) to the diagnosis of typical articular manifestations of the disease, along with an assessment that involved cardiology, ophthalmology, physical-therapy specialties; performing at the same time a coagulation assay and peripheral blood smear.
Results: no sex prevalence was found and family history was present in more than the half of patients, where maternal line prevailed. Hemorrhagic familial trend was positive in 36 cases. The main manifestations found in these patients were described to this health condition, the existence of mucous-cutaneous bleeding predominated or as a consequence of invasive procedures in 181 children; which was associated with the presence of disorders associated with macro-platelets and the finding of disperse platelets in peripheral-blood smears.
Conclusions: the results evidenced the presence of qualitative platelet disorders that must be evaluated by means of much more specific coagulation assays, not in existence in the province, which must include aggregation and platelet function.
DeCS: Type-III Ehlers–Danlos Syndrome; Blood coagulation disorders.
INTRODUCCIÓN
La anomalía básica en el síndrome de Ehlers-Danlos es la formación anormal del tejido conectivo que constituye la armazón que da soporte y sostén a la mayoría de los órganos y sistemas.1 La movilidad de una articulación está determinada por los ligamentos que la controlan, por lo que la causa primaria de la hiperlaxitud articular es ligamentosa y ello está definido por los genes que codifican la síntesis de colágeno, elastina y fibrilina. En la población normal la hiperlaxitud es máxima al nacimiento y disminuye de forma cada vez más lenta durante el transcurso de la vida. La hipermovilidad articular es más frecuente en mujeres que en hombres y aparece en alrededor de un 10% en caucásicos.2 Sólo cuando la hipermovilidad e hiperextensión de las articulaciones producen síntomas se considera que la persona tiene un "síndrome de hiperlaxitud". En el contexto de la hiperlaxitud articular, las enfermedades genéticas como el síndrome de Marfan, la osteogénesis imperfecta, y el síndrome de Ehlers-Danlos constituyen un grupo particular. Este último debe sospecharse clínicamente. Se manifiesta por hipermovilidad articular con hiperextensibilidad asociada a anomalías cutáneas. La piel es frágil e hiperelástica por lo que se daña con facilidad y presenta dificultades para la cicatrización. La tendencia a las luxaciones es típica del trastorno así como la fragilidad de los vasos sanguíneos.3
En 1997 se realizó la última clasificación del síndrome de Ehlers-Danlos por un grupo de expertos en Villefrance, Francia4, que incluyó 6 grupos principales siendo los más frecuentes el I y el III. Los tipos I y II (clásica) con anomalía del colágeno tipo V, tipo III (hipermovilidad) sin anomalía definida, tipo IV (vascular) con anomalía del colágeno tipo III, tipo VI (cifoescoliosis) debida a deficiencia de lisil-hidroxilasa y tipo VII que incluye la artroclasia (deficiencia de cadenas del colágeno tipo I), dermatopraxis (deficiencia de enzima del colágeno tipo I) y otras variantes menos frecuentes. Todas son autonómicas dominantes, excepto la dermatopraxis y cifoescoliosis, que son recesivas. En pacientes con síndrome de hipermovilidad articular generalizada (síndrome de Ehlers-Danlos tipo III), Child5describió una alteración en la proporción del colágeno tipo I con el tipo III semejante a lo observado para la osteogénesis imperfecta y la microscopía electrónica mostró fibras colágenas de menor diámetro; Hermanns-Le6 encontró anomalías en el diámetro y la distribución de las fiabas de colágeno. A pesar de esto, todavía la alteración bioquímica precisa y el defecto genético en esta variante del síndrome no son bien conocidos. Estudios recientes indican un probable rol de los defectos del gen de Tenascina-X7 y un defecto del gen Col5A3 evidenciado en 13 personas8. Ambos estudios necesitan ser ampliados.
La alta frecuencia de esta enfermedad genética unido a la falta de conocimiento de que puede causar daño en múltiples sistemas orgánicos, el hecho que el cuadro clínico no sea dramático, no haya signos inflamatorios, no existan exámenes de laboratorio ni signos radiológicos específicos asociado a que existe la sensación de que el tratamiento no es efectivo, hace que los médicos no se interesen por esta afección originando un sub-registro de la enfermedad unido a la frecuencia con que se remiten a consulta de hematología pacientes con síndrome de Ehlers-Danlos que presentan tendencia al sangramiento cutáneo a pesar de que no ha sido reportada la existencia de trastornos hemostáticos como parte de la clínica de la enfermedad9, nos motivó a caracterizar clínicamente este síndrome con el objetivo de definir un diagnóstico precoz, seguimiento y tratamiento en las distintas instancias de salud, así como evaluar si existe un defecto de la coagulación específico que forme parte de la enfermedad.
MATERIAL Y MÉTODO
Se realizó un estudio descriptivo y transversal de 305 niños entre 5 y 18 años con hipermovilidad articular atendidos en el Hospital Pediátrico Docente "Pepe Portilla" entre enero de 2010 y junio de 2011. Se consideraron como criterios mayores para el diagnóstico la existencia de hipermovilidad articular generalizada y afectación cutánea variable y menores la historia de luxaciones articulares recurrentes, dolor articular crónico o antecedentes familiares.Mediante el interrogatorio de cada paciente y/o de la persona acompañante se identificó la edad, género e historia familiar sugestiva de la enfermedad.
Se realizó el examen físico completo y la evaluación de la movilidad articular según los criterios de Beighton4 para el diagnóstico de síndrome de Ehlers-Danlos tipo III. La hiperextensibilidad cutánea, piel "gomosa, aterciopelada o blanda" se definió según la apreciación y experiencia del examinador. Se definió como síndrome de Ehlers-Danlos tipo III la existencia de hipermovilidad articular asociada a otro signo mayor o al menos un signo menor. Se identificaron las alteraciones ortopédicas y otros elementos clínicos propios de la enfermedad en cada uno de los pacientes.
Disponiendo del consentimiento informado de los pacientes y/o su tutor se realizó además coagulograma completo utilizando coagulómetro de cuatro canales marca Stara de la Diagnostica Otago y extendido de sangre periférica para determinar la existencia de alteraciones de la coagulación no diagnosticadas previamente.
RESULTADOS
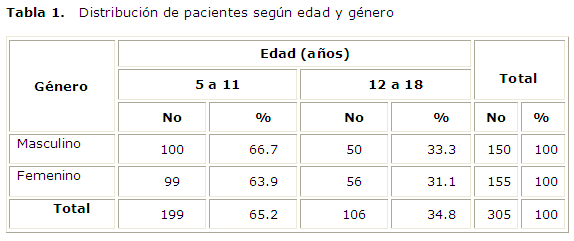
En la tabla 1 se observa que en los pacientes incluidos en el estudio no hubo predominio de género y la mayor cantidad de ellos pertenecieron a los de edades más tempranas.
Los antecedentes patológicos familiares de la enfermedad estuvieron presentes en 223 pacientes, predominando la línea materna que constituyó el 48,2% de los niños con historia familiar positiva (Tabla 2 ).

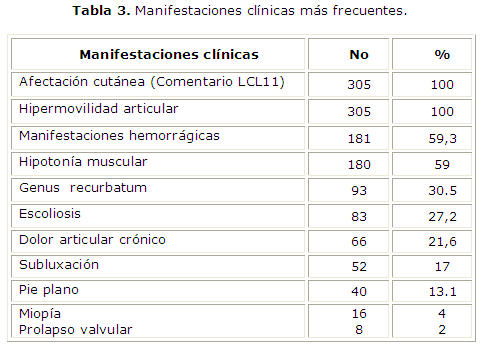
El comportamiento clínico de los enfermos mostró que los síntomas articulares y cutáneos estuvieron presentes en todos ellos (Tabla 3 ). La afectación cutánea e hipermovilidad articular fueron universal y las manifestaciones hemorrágicas ocuparon un lugar destacado en las manifestaciones clínicas de los pacientes.
El estudio de la lámina periférica evidenció la presencia de macroplaquetas, plaquetas dispersas y escasa formación de grumos en el 62 % de los pacientes, siendo normales todos los demás parámetros del coagulograma.
DISCUSIÓN
Los pacientes con los diversos sub-tipos de síndrome Ehlers-Danlos comparten manifestaciones clínicas incluida la hipermovilidad articular que es la principal característica del tipo III10. Debido a las alteraciones genéticas que afectan la síntesis del colágeno, la hiperelasticidad de otras estructuras del organismo, en particular la piel, es un elemento que usualmente puede ser identificado al examen del paciente.11 El estudio realizado no evidenció predominio de género como ha sido señalado en la literatura sobre el tema12 probablemente por el hecho de que las series son de menor número de pacientes que incluyen niños y adultos. La mayor parte de los pacientes evaluados fueron niños menores de 11 años, en los que se conoce que la hipermovilidad es más frecuente, la cual puede incluso mejorar con el paso de los años. Los antecedentes familiares fueron predominantemente en la línea materna y fue inusual en ambos progenitores asociados. A pesar de la herencia autosómica dominante, la ausencia de antecedentes familiares conocidos en un grupo de los pacientes es, como se ha señalado, el resultado del no reconocimiento de enfermos con manifestaciones leves, no identificadas o incorrectamente interpretadas.13 En los familiares afectados, el dolor articular crónico fue la manifestación más referida, lo cual se corresponde con lo reportado acerca de que este síntoma es el más frecuente en los adultos con el síndrome y, de hecho, una de las causas más usuales de dolor reumatológico en la clínica de adultos.14 Como es de esperar, la hipermovilidad articular también fue encontrada en una gran cantidad de los familiares afectados y más del 20% tenían historia de luxaciones articulares, muchos de ellos recidivantes.
Como es característico de este sub-tipo del síndrome15, las manifestaciones cutáneas se encontraron en la totalidad de los enfermos estudiados. Éstas fueron ligeras y predominó la piel blanda, hiperelástica y aterciopelada, sin evidencia de pseudotumores u otras anomalías dermatológicas, que son típicas del síndrome de Ehlers-Danlos tipo I.16 La hipermovilidad articular estuvo presente en todos los niños estudiados asociada en más de la mitad de ellos a hipotonía muscular evidenciable al examen físico y sintomática en la mayoría. Otros hallazgos articulares como el Genus recurvatum, escoliosis y pie plano fueron también encontrados, lo que coincide con lo descrito en esta enfermedad.2, 8
Es de señalar que 52 pacientes tenían historia de subluxación recidivante, lo que indica el daño articular temprano. La existencia de dolor crónico, aunque menos frecuente que en los adultos, es una manifestación que requiere suficiente atención para lograr disminuir las limitaciones que con el paso de los años pueden producirse en estos pacientes. Los prolapsos valvulares no fueron frecuentemente encontrados y en todos los casos su presencia no evidenció repercusión identificable en la función cardiovascular como es típico de esta variante de la enfermedad, en la que las alteraciones cardiovasculares, de existir, son ligeras.16
El examen oftalmológico debe realizarse de forma regular, ya que es frecuente la miopía en algunas de las formas clínicas del síndrome3 así como el estrabismo y las escleras celestes. La evaluación realizada sólo evidenció la existencia de miopía no acompañada de otras alteraciones oftalmológicas. Los resultados obtenidos en el coagulograma no evidenciaron alteraciones dependientes de defectos plasmáticos lo que coincide con lo reportado en la literatura.18
El recuento plaquetario fue normal en todos los casos y es de resaltar que tanto la prueba del lazo como el tiempo de sangramiento, estudios que pueden ser positivos en presencia de alteraciones vasculares o plaquetarias fueron normales. El examen de la lámina periférica mostró la presencia de macroplaquetas, plaquetas dispersas y escasa formación de grumos en mas de la mitad de los pacientes, lo que sugirió la posibilidad de un trastorno cualitativo plaquetario responsable de las manifestaciones hemorrágicas referidas sólo o combinada con el daño de la pared vascular que caracteriza a las distintas variantes del síndrome en mayor o menor cuantía. Sólo en algunas investigaciones ha sido mostrada la existencia de alteraciones cualitativas plaquetarias en pacientes con otras variantes del síndrome.17, 18 Los resultados obtenidos en la presente investigación podría ser un marcador del síndrome de Ehlers Danlos tipo III y el estudio detallado del mismo requiere investigaciones posteriores.
Usualmente se diagnostican en estos pacientes problemas puntuales como tendinitis y bursitis, y si se reconoce la hiperlaxitud, no se aprecia su significado ni sus posibles complicaciones por lo que la caracterización de la enfermedad constituye un objetivo primordial que permitirá la evaluación y el seguimiento de estos enfermos para lograr en ellos una calidad de vida aceptable disminuyendo, de ser posible, sus manifestaciones crónicas y las limitaciones que ellas producen mediante una intervención temprana y adecuada de la acción médica. La identificación de una potencial anomalía funcional plaquetaria en ellos constituye un hallazgo que requiere de estudios posteriores para su identificación precisa.
REFERENCIAS BIBLIOGRÁFICAS
1. WW Tan P, Song C, Lalonde D. Ehlers-Danlos syndrome associated with cleft lip and palate. Can J Plast Surg. 2009[cited dec.2012]; 17(4): e24-e26. Available from: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2827283/
2. Bravo Jaime F. Síndrome de Ehlers-Danlos con especial énfasis en el síndrome de hiperlaxitud articular. Rev. méd. Chile [revista en la Internet]. 2009 Nov [citado 2013 Abr 09]; 137(11): 1488-1497. Disponible en: http://www.scielo.cl/scielo.php?script=sci_arttext&pid=S0034-98872009001100013&lng=es
3. Bravo J. Significado e importancia de estudiar a las personas con hiperlaxitud articular. Rev Chil reumatol 2008;24:4-5.
4. Beighton P, De Paepe A, Steinmann B, Tsipouras P, Wenstrup RJ. Ehlers-Danlos Syndromes: Revised Nosology, Villefranche, 1997. Am J Med Genet[internet]. 1998 Apr 28[cited nov. 2012]; 77(1):31-7. Available from: http://prettyill.com/downloads/Ehlers-Danlos_Syndrome_Revised_Nosology.pdf
5. Child AH. Joint Hypermobility Syndrome: inherited disorder of collagen synthesis. J Rheumatol 1986; 13(2): 239-42.
6. Hermanns-Le T, Pierard GE. Ultra structural alterations of elastic fibers and other dermal components in Ehlers-Danlos syndrome of the hypermobile type. Ann J Dermatopathol[internet] 2007[cited nov. 2012]; 29(4):370-3. Available from: http://journals.lww.com/pages/login.aspx?ContextUrl=%2famjdermatopathology%2fsecure%2fpages%2fpurchase.aspx%3fan%3d00000372-200708000-00007
7. Zweers MC, Hakim AJ, Grahame R, Schalkwiik J. Joint hypermobility syndromes: the pathophysiologic role of Tenascin-X gene defects [review]. Arthritis Rheum[internet] 2004[cited nov. 2012]; 50(9): 2742-9. Available from: http://onlinelibrary.wiley.com/doi/10.1002/art.20488/pdf
8. Levy H. Síndrome de Ehlers Danlos tipo hiperlaxitud. Revisión (Sed tipo III, Síndorme de Ehlers Danlos tipo III. Asociación Síndromes de Ehlers Danlos e Hiperlaxitud 2001-2010 (actualizado 2011 Ene 25; citado 2011 Feb 20). Disponible en: https://sites.google.com/site/rededargentina/sindrome-de-ehlers-danlos-tipo-hiperlaxitud
9. Yenicesu I, Ucan D, Soysal A, Büyükasik Y, Gümrük F. Platelet reliase defect in a child with Ehlers-Danlos syndrome. Pediatr Hematol Oncol[internet] 2000[cited nov. 2012];17(2):193-4. Available from: http://informahealthcare.com/doi/pdf/10.1080/088800100276587
10. Prockop D J, Czarny-Ralajczak M. Disorders of connective tissue. In: Harrison's Principles of Internal Medicine. 17th edition. Tomo II. New York: Mc Graw Hill; 2008: 2461-8.
11. Watanabe A, Kosho T, Wada T, Sakai N, Fujimoto M, Fukushima Y, Shimada T. Genetic Aspects of the Vascular Type of Ehlers-Danlos Syndrome. Circ J[internet] 2007[cited nov. 2012];71(2): 261-265. Available from: https://www.jstage.jst.go.jp/article/circj/71/2/71_2_261/article
12. Miranda-Gómez A. Hiperelasticidad cutánea e hiperlaxitud articular. Rev Mex Dermatología[internet] 2008[citado nov. 2012];52(3): 248-55. Disponible en: http://www.nietoeditores.com.mx/download/Dermatologia/mayo-junio2008/Derma2008-52%283%29111-20.pdf
13. Tofts LJ, Elliott EJ, Munns C, Pacey V, Sillence DO. The differential diagnosis of children with joint hypermobility: review of the literature. Pediatric Rheumatology[internet] 2009[cited nov. 2012];7:1-10. Available from: http://www.ped-rheum.com/content/7/1/1
14. Keer H, Elsevier G. Hipermobility, fibromyalgia and chronic pain. Reumatology 2010;26:194-202.
15. EUA. Asociación Síndromes de Ehlers Danlos e Hiperlaxitud. Tratamiento del Síndrome de Ehlers Danlos. [Internet]. 2010 Mar 14. Disponible en: http://asedh.org/quesh.php [citado 2011 Ene 24]
16. McDonnell NB, Gorman BL, Mandel KW, Schurman SH, Assanah-Carroll A, Mayer SA, Najjar SS, Francomano CA. Echocardiographicfindings in classical and hypermobile Ehlers-Danlos syndromes. Am J Med Genet[internet] 2006[citado nov. 2012];140(2):129-36. Available from: http://onlinelibrary.wiley.com/doi/10.1002/ajmg.a.31035/pdf
17. Gibbins JM. Platelet adhesion signalling and the regulation of thrombus formation. Journal of Cell Science[internet] 2004[cited nov. 2012];117:3415-25 . Available from: http://jcs.biologists.org/content/117/16/3415.long
18. Onel D, Ulutin S B, Ulutin O N. Platelet defect in a case of Ehlers-Danlos syndrome. Acta Haematol 1973;50:238-44.
Recibido: 12 de noviembre del 2012.
Aprobado: 3 de abril del 2013.
Dra. Mirta Caridad Campo Díaz. Especialista de Segundo Grado en Hematología y Pediatría. Profesora Auxiliar y Consultante. Investigadora Auxiliar. Máster en Atención Integral al Niño. Hospital Provincial Pediátrico Docente “Pepe Portilla”. Pinar del Río. Correo electrónico: mcampo@princesa.pri.sld.cu
