










Meu SciELO
 Serviços customizados
Serviços customizadosServiços Personalizados
Journal

Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkRevista Médica Electrónica
versão On-line ISSN 1684-1824
Rev. Med. Electrón. v.31 n.1 Matanzas jan.-fev. 2009
POLICLÍNICO DOCENTE DR. RAMÓN MARTÍNEZ. VARADERO.
Asesoramiento genético preconcepcional a familia con hijo afectado de Distrofia Muscular de Duchenne
Preconception Genetic Management to a family with a child affected by Duchenne Muscular Dystrophy
AUTORAS
Dra. Hilda Álvarez de la Campa Gil. (1)
E-mail: genetica.mtz@infomed.sld.cu
Dra. Milaydi Moreira González. (1)
Dra. Dayri Falcón Rodríguez. (2)
Lic. Claribel Lugo Rodríguez.(3)(1)Especialista de 1er. Grado en Medicina General Integral. Máster en Asesoramiento Genético.Policlínico Docente " Dr. Ramón Martínez". Varadero.
(2)Especialista de 1er. Grado en Genética Clínica. Profesora Instructora. Centro Provincial de Genética.Matanzas.
(3)Licenciada en Enfermería. Máster en Asesoramiento Genético. Policlínico Docente " Dr.Ramón Martínez".VaraderoRESUMEN
Las personas con discapacidad representan alrededor del 10 % de la población, por ello su atención va desde su prevención, mejorar su calidad de vida y la integración social. De estas discapacidades una de las más frecuentes en aparecer son las físico-motoras y causa de éstas son las Distrofias Musculares, por lo que en este trabajo se realizó una revisión bibliográfica actualizada sobre la Distrofia Muscular de Duchenne previa identificación de un caso afectado en una familia del consejo popular Boca de Camarioca del municipio de Varadero, y se propuso una metodología para el Asesoramiento Genético Preconcepcional de esta enfermedad, dado que el afectado tiene dos hermanas en edad reproductiva. Se tomaron como base los principios básicos de este proceso mostrando así la interrelación dinámica entre sus elementos y se elaboró un documento educativo-informativo para una mayor comprensión de la enfermedad y que pueda ser discutido en el seno de la familia.
DeCS:
DISTROFIA MUSCULAR DE DUCHENNE/genética
DISTROFIA MUSCULAR DE DUCHENNE/prevención & control
ATENCIÓN PRECONCEPTIVA/métodos
EDUCACIÓN AL PACIENTE COMO ASUNTO
CALIDAD DE VIDA
HUMANOSINTRODUCCIÓN
Las personas con discapacidad representan alrededor del 10 % de la población, por ello su atención va desde su prevención, mejorar su calidad de vida y la integración social. De estas discapacidades una de las más frecuentes en aparecer son las físico-motoras y causa de éstas son las Distrofias Musculares. Éstas son enfermedades hereditarias caracterizadas por degeneración del músculo esquelético o voluntario, que controlan el movimiento y se originan por una mutación genética, por lo que afectan a uno o varios miembros de la familia. Su curso clínico es progresivo, se presentan a cualquier edad y existen varias formas clínicas que se diferencian unas de otras por el patrón de herencia, el nivel de severidad, edad de inicio de la enfermedad, por la distribución de los grupos musculares afectados y más frecuentemente el paso al que avanzan los síntomas. En todas las formas se detectan anormalidades microscópicas en el examen histológico del músculo estriado y no suelen producirse remisiones. (1-4)
La Distrofia Muscular de Duchenne (DMD) es una enfermedad recesiva ligada al X, debido a mutaciones alélicas ocurridas en el gen que codifica para la Distrofina, cuya ausencia causa degenaración muscular progresiva. El gen responsable está localizado en el brazo corto del cromosoma X (región Xp21). Es el más grande y complejo hasta ahora conocido, con 2 500 kb de longitud y con cerca de 79 exones que representan menos de 1% de la longitud total del gen. (5) Una característica del gen es que la deleción es el defecto génico más frecuente, con incidencia de hasta el 70 % en algunas poblaciones estudiadas (6). Estas deleciones son muy heterogéneas, tanto en su ubicación como en su longitud, algunas involucran a todo el gen, mientras que en otros casos se trata de una deleción parcial de tamaño variable. (7)
Se considera la forma infantil la más severa y letal, por no disponer en la actualidad de un tratamiento curativo. Su incidencia oscila entre 1:3500-1:4854 nacimientos (8) y su comienzo ocurre temprano en la vida, aunque raramente se da un diagnóstico antes de los 4 años. Puede presentarse al nacimiento con hipotonía, fallo del crecimiento y retardo del desarrollo para caminar. Otros signos clínicos son debilidad muscular simétrica y bilateral, marcha anadeante, hiperlordosis, debilidad de las rodillas y caderas (signo de Gower) y tendencia a caminar con la punta de los dedos. La pseudohipertrofia muscular se presenta en algunos músculos (gemelos, deltoides, etc.) y se debe a exceso de tejidos conectivo y adiposo y es frecuente que se necesite una silla de ruedas a los 12 años. Según progresa la enfermedad se afecta la musculatura lisa, pudiendo presentarse una miocardiopatía dilatada. Las alteraciones electrocardiográficas son evidentes en la adolescencia y en algunos casos existen trastornos de la motilidad intestinal. El cociente de inteligencia puede estar disminuido en el 70 % y puede presentarse hiperplasia del timo, hiperestroginemia y retardo de la pubertad. La respiración se afecta en las etapas tardías llevando a infecciones respiratorias que marcan el final de la enfermedad la que ocurre entre los 20 y 30 años de edad. La concentración de la enzima creatinina quinasa se encuentra elevada en suero y otros estudios que ayudan al diagnóstico son: la electromiografía, la biopsia de músculo, el Western Blot y el análisis inmunohistoquímico. (6,9,10)
En el consejo popular Boca de Camarioca, del municipio de Varadero, se identifica una familia con un integrante afectado de DMD por lo que se propone una metodología para el Asesoramiento Genético (AG) Preconcepcional de esta enfermedad.MÉTODO
Se elaboró una propuesta metodológica de Asesoramiento Genético Preconcepcional, dirigido a una familia identificada en el consejo popular Boca de Camarioca del municipio de Varadero con un miembro afectado de DMD el cual tiene dos hermanas en edad reproductiva sin ninguna orientación genética.
PRESENTACIÓN DEL CASO
Niño afectado de 15 años del sexo masculino, raza blanca, que fue remitido a los 6 años al servicio de neurología por presentar dificultad para la marcha y correr, lo cual se acentuaba con el paso de los años, además de hipertrofia marcada de los gemelos, microcefalia y alteraciones electroencefalográficas. El niño ya no camina. Antecedentes de: embarazo fisiológico, parto distócico (forceps), llanto demorado y cianosis. Madre fallecida (leucemia) y hermana con retraso mental ligero.
Datos positivos al examen físico: Microcefalia, fascie de cusching (por tratamiento con esteroide), puente nasal deprimido, cuello corto, tórax: troncular, extremidades inferiores con atrofia de los músculos, debilidad muscular proximal, pensamiento lento, (retraso mental moderado, que se debe a traumas del periparto, ya que en la literatura revisada sólo llegan a presentar retraso mental ligero el 70 % de los casos.
Complementarios realizados:CPK: 1254 (elevado), electromiografía positiva con patrón miopático. Exploración electrofisiológica del reflejo Aquileano como parte de la caracterización del estado evolutivo de la enfermedad y Biopsia de muslo: Positiva. Se confeccionó la historia clínica genética haciendo un interrogatorio exhaustivo sobre antecedentes patológicos familiares y se realizó el árbol genealógico familiar a partir de la información obtenida.
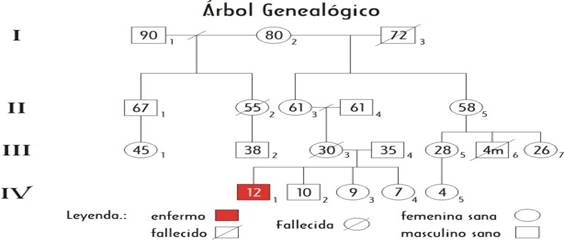
II- METODOLOGÍA QUE SE PROPONE:
Se propone el Asesoramiento Genético en 3 sesiones de trabajo y se utilizó la entrevista como instrumento fundamental, lo que permitió la confección de la historia clínica genética y el árbol genealógico que ayudó a conducir con mayor flexibilidad a la familia durante el Asesoramiento. Se evaluó en cada sesión la satisfacción de las necesidades individuales acerca de la información brindada. Los aspectos discutidos en las sesiones se planificaron de la siguiente forma:
PRIMERA SESIÓN:
• Iniciar el proceso de empatía con la pareja.
• Ofrecer información general sobre la enfermedad.
• Confeccionar historia clínica y árbol genealógico.
• Indicar exámenes complementarios.
• Conocer inquietudes, pensamientos y creencias que poseen los pacientes.
SEGUNDA SESIÖN:
• Entregar los resultados de los exámenes realizados.
• Profundizar en el conocimiento de la enfermedad.
• Mantener apoyo psicológico y afianzarlo.
• Brindar opciones reproductivas.
• Entregar hoja educativo-informativa.
• Invitar a familiares de los pacientes a la próxima consulta .
TERCERA SESIÖN:
• Evaluar y explorar lo abordado en la consulta anterior.
• Valorar grado de comprensión de la familia sobre la información ofrecida.
• Esclarecer dudas para disminuir temores, ansiedad, preocupaciones, así como los sentimientos de culpa.
• Continuar el apoyo psicológico.
• Brindar seguimiento por nuestro servicio.
III- Elaboración de una hoja educativo-informativa .
Se elaboró una hoja educativo-informativa que resume los propósitos fundamentales.
DISCUSIÓN
El AG preconcepcional tiene como premisa fundamental alcanzar una plena salud reproductiva en las personas, éste debe preceder siempre al prenatal evitando así un diagnóstico postnatal de la enfermedad, con el objetivo de disminuir la frecuencia de estos nacimientos. Se establece el motivo de visita al hogar, el cual consiste en brindar AG a una familia donde existe un miembro afectado de DMD y varios familiares del sexo femenino. Se saluda a los presentes, se presenta el equipo de trabajo y se crea un ambiente psicológico adecuado que garantiza el desarrollo de una buena comunicación e invitar a una cooperación recíproca. Se realiza la entrevista a través de un interrogatorio detallado donde se adquieren los datos necesarios para confirmar el diagnóstico: éste incluye el interrogatorio a los abuelos, el examen físico al paciente y como dato relevante la historia clínica del afectado, donde se recoge su evolución y los complementarios realizados. Se confecciona la historia clínica genética analizándose hasta 4 generaciones donde existen mujeres que se encuentran en edad reproductiva y cuyo AG preconcepcional está dirigido a ofrecer opciones antes del matrimonio o antes de la concepción, con el objetivo de prevenir las enfermedades hereditarias . Se elabora el árbol genealógico para determinar un posible patrón de herencia y el cálculo posterior del riesgo en la familia y se comenta sobre características de la enfermedad. Se da a conocer a los familiares que existen estudios para determinar el estado de portador de la enfermedad a través de pruebas de laboratorio previo consentimiento informado y las consecuencias que traería para la familia la no realización de los mismos. Los resultados incluye varias posibilidades.
Primero: que el estudio molecular precise la mutación en el caso del afectado y, por tanto, se pueda realizar estudio de portadoras a las mujeres en edad fértil.
Segundo: que el estudio molecular no precise la mutación por estudio directo en el caso del afectado y, por tanto, el estudio de portadoras a las mujeres en edad fértil sería por estudios de análisis de ligamiento. Se ofrece apoyo psicológico mostrando confianza, atención y comprensión ante sus inquietudes y preocupaciones con el objetivo de preparar adecuadamente a la familia y disminuir la ansiedad ante los posibles resultados de los exámenes a realizar, se programa la próxima consulta en la cual se continúa con la comunicación como elemento vital en la comprensión de la información y se procede a ofrecer los resultados de los exámenes realizados, donde el afectado no presenta ninguna de las mutaciones que se estudian en el país, lo que no excluye el diagnóstico de DMD, pues el paciente desde el punto de vista clínico y de complementarios realizados anteriormente nos lleva a pensar en esta enfermedad, por tanto, se orienta que los exámenes de las hermanas se realizará por estudios indirectos de marcadores. Se explica nuevamente sobre las manifestaciones clínicas fundamentales de la enfermedad con lenguaje claro y sencillo, aclarando todas las dudas que surjan y se informa con relación al pronóstico de la misma cuya forma infantil se considera la más severa y letal, por no disponer en la actualidad de un tratamiento curativo. También se explica sobre el patrón de herencia (enfermedad recesiva ligada al cromosoma X), es decir, que la enfermedad debe transmitirse de mujeres portadoras a sus hijos varones, por tanto, existe la probabilidad de que tengan un hijo con la misma enfermedad de su hermano. Si los estudios indirectos para las hermanas fueran positivos (portadoras) éstas tendrán la oportunidad de realizarse diagnóstico prenatal de la DM, para lo que es necesario realizar el cariotipo fetal determinando el sexo del feto (la toma de la muestra puede realizar a través de la biopsia coriónica entre las 10-12 semanas de gestación o mediante la amniocéntesis entre las 16-18 semanas de gestación). Si el sexo del feto es femenino se le explica que existe un 50 % de que sus hijas sean portadoras como ellas para cada embarazo y si el feto es masculino se procede entonces a realizar el análisis de la mutación en el ADN de las células fetales, que en caso de ser positivo se le daría la opción de interrumpir el embarazo o prepararse para el nacimiento de un niño enfermo. Si los estudios indirectos no fueran concluyentes, se ofrecería el estudio de sexo, con posibilidad de interrumpir los varones por la probabilidad de ser afectados. Se evalúa el grado de comprensión de la familia, así como sus expectativas y dudas surgidas además de continuar el seguimiento periódico por las especialidades médicas (Cardiología, Oftalmología, Fisiatría, MGI ) ya que son de vital importancia en la prevención de complicaciones, ayudando de esta forma a mejorar la calidad de vida del enfermo. Se continúa con apoyo psicológico y se entrega hoja informativa con la información brindada. Se sugiere para la próxima consulta la participación de algún familiar interesado, principalmente aquellas personas en edad reproductiva y orientar sobre dudas acerca de la información que se brindó con anterioridad y la necesidad de continuar en contacto con los servicios de genética para esclarecer las frustraciones y conflictos que puedan surgir en los miembros de la familia como consecuencia de la información recibida, lo que generan angustia, depresión, ansiedad y otros estados psicológicos en la familia. El seguimiento estará en correspondencia con las necesidades del caso y se comunicará asistir tantas veces lo necesiten a los centros de referencia, sobre todo aquéllos en edades reproductivas, permitiendo que se pueda evaluar en un futuro las implicaciones genéticas de la historia familiar de la enfermedad y que exista un acercamiento del médico de familia, el asesor genético y el genetista para futuras investigaciones. Informar a los pacientes los centros de referencias donde podrán dirigirse para la atención y seguimiento:
-Consultas de salud reproductivas disponibles en todos los policlínicos ofrecidas por un máster en AG o MGI capacitado al efecto.
-Servicios de genética municipales o provinciales
REFERENCIAS BIBLIOGRÁFICAS
1. Colectivo de Autores: Por la vida. 2da ed. La Habana: Editora Abril; 2003.
2. Tucson A. Datos sobre la Distrofia. Sociedad Mexicana de la Distrofia Muscular; 2001.
3. Hidalgo PC. Bases Moleculares de las Distrofias Musculares Ligadas al Cromosoma x: Duchenne y Becker. Inicio de una nueva época en la Medicina. Medicentro. 1989; 4(2): 254-7.
4. Koening M, Hoffman Ep, Bertelson CJ, Monaco AP, Feener C, Kunkel LM. Complete Cloning of the Duchenne Muscular distrophy(DMD) c DNA and Preliminary Genomic Organization of the DMD gene is normal and affected individuals. Cell. 1987; 50: 509-17.
5. Hoffman EP, Brown RH, Kukel LM. Dystrophin the protein product of the Duchenne muscular Dystrophy locus. Cell. 1987; 51: 919-28.
6. Matthews J. Muscular Dystrophy Campaign. London; 2002.
7. David G, Brooks MD. División of medical Genética. Philadelphia, PA:University of Pennsylvania Medical Center; 2003.
8. Adams RD. Principles of Neurology. 6th ed. New York: McGraw-Hill; 1997.p. 415-7.
9. Douglas R, Stewart MD. Division of Medical Genetics.Philadelphia PA:University of Pennsylvania Medical Center;2004.
10. Kenneth LT. Origins and early description of Duchenne muscular Dystrophy. Muscle Nerve. 2003; 28: 402-22.
11. Cervantes DA. Distrofia muscular.México:Instituto Nacional de Rehabilitación;2005.
12. Mueller F, Young D. Asesoramiento Genético. 10ed.España:Emery's, Marbán; 2001.p.243-73.
13. Rodríguez M. Diagnóstico de DMD mediante análisis del ácido desoxirribonucleico y su aplicación en la prevención. Rev Cubana Pediatr.1996;68(1): 10-20.SUMMARY
Disabled people represent around 10 % of the population. Their attention covers prevention, life quality improvement, and social integration. One of the most frequent disabilities is the physic-motor one and muscular dystrophies are their cause. To make this work we carried out an up-dated bibliographic review on Duchenne muscular dystrophy after being identified a case in a family living in the popular council Boca de Camarioca, municipality of Varadero. We proposed a methodology for the Preconception Genetic Management of this disease, taking into account that the patient has two sisters in reproductive age. As a base we took the main principles of this process, showing the dynamic interrelation between its elements and elaborated an informative-educative document for a better comprehension of the disease to be discussed by the members of the family.
MeSH:
MUSCULAR DYSTROPHY, DUCHENNE/genetics
MUSCULAR DYSTROPHY, DUCHENNE/prevention & control
PRECONCEPTION CARE/methods
PATIENT EDUCATION AS TOPIC
QUALITY OF LIFE
HUMANSCÓMO CITAR ESTE ARTÍCULO
Álvarez de la Campa H , Moreira González M, Falcón Rodríguea D,Lugo C. Asesoramiento Genético Preconcepcional a familia con hijo afectado de Distrofia Muscular de Duchanne. Rev méd electrón[Seriada en línea] 2009; 31(1). Disponible en URL: http://www.revmatanzas.sld.cu/revista%20médica/ano%202009/http://www.revmatanzas.sld.cu/revista%20medica/ano%202009/vol1%202009/tema11.htmvol1%202009/tema11.htm [consulta: fecha de acceso]

