










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Cirugía
versión impresa ISSN 0034-7493versión On-line ISSN 1561-2945
Rev Cubana Cir vol.56 no.3 Ciudad de la Habana jul.-set. 2017
PRESENTACIÓN DE CASO
Neurofibromatosis sin neurofibromas con tumor mediastinal como forma de debut
Neurofibromatosis without neurofibromas. Case report with a mediastinal tumor as the first sign of the disease
Nélida Ramos Díaz, Radamés Isaac Adefna Pérez, Françoise T. Izquierdo Lara, Yunior Luis Pulido Prieto, Noralys Lara Fernández
Hospital Clínico Quirúrgico Docente "Miguel Enríquez". La Habana, Cuba.
RESUMEN
Se presenta paciente con historia familiar de neurofibromatosis (NF) tipo I, con síntomas y signos sugestivos de esta enfermedad que refiere dolor torácico paravertebral izquierdo al cual mediante estudios de imagen se le diagnostica lesión tumoral en mediastino posterior. Es de notar la presencia de manchas cutáneas características de la neurofibromatosis, localizadas solo en un dermatoma del cuerpo sin neurofibromas en ninguna otra localización. Se intervino quirúrgicamente el enfermo, se resecó una gran masa mediastinal que se confirmó histológicamente ser un neurofibroma. La evolución posquirúrgica fue satisfactoria. Se discuten las singularidades de este enfermo sobre la base de criterios diagnósticos de neurofibromatosis tipo I, pero sin neurofibromas periféricos hasta ese momento. La posibilidad de una neurofibromatosis segmentaria tampoco se descarta. Se hace énfasis en la necesidad de resección de cualquier lesión tumoral en el contexto de este síndrome genético por la frecuencia de lesiones malignas asociadas a la neurofibromatosis y a la progresión hacia la malignidad de lesiones primariamente benignas.
Palabras clave: neurofibromatosis; enfermedad de Von Recklinghausen; neurofibroma; mediastino posterior; tumores neurogénicos; neurofibromatosis segmentaria.
ABSTRACT
A patient with a family history of Neurofibromatosis type I was presented. The patient referred symptoms and signs suggestive of that disease and complaint of left paravertebral chest pain. Imaging investigations were done and a posterior mediastinal tumor was diagnosed. It is important to highlight the presence of characteristics spot of neurofibromatosis localized only in one dermatome without neurofibromas in any other part of the body. The patient underwent a surgical intervention and a left thoracotomy was done, a large posterior mediastinal tumor was found and totally resected. The histology confirmed a neurofibroma. The postoperative evolution was satisfactory. The singularities of this patient were discussed, especially diagnosis criteria for neurofibromatosis type I, and the lack of peripheral neurofibromas until that moment. The possibility for a segmental neurofibromatosis also was considered. We pointed out about the necessity to remove any neoplastic lesion in the background of this genetic syndrome due to the high frequency of malignancies associated with neurofibromatosis, and also related with the malignant degeneration these tumors can develop.
Keywords: neurofibromatosis, Von Recklinghausen disease, neurofibroma, posterior mediastinum, neurogenic tumors, segmental neurofibromatosis.
INTRODUCCIÓN
Las neurofibromatosis son enfermedades genéticas del sistema nervioso que afectan el desarrollo y crecimiento de los tejidos de las células neurales. Existen dos variantes denominadas tipo 1 y tipo 2 caracterizadas por una mutación en el cromosoma 17 y 22 respectivamente.1 Estas afecciones causan tumores en los nervios, además de cambios en la piel y deformidades óseas. La NF tipo I o enfermedad de Von Recklinghausen, representa el 95 % de todos los casos. Se presenta con un espectro amplio de expresión fenotípica y evolución impredecible. Se estima que solo un 5 % de los pacientes van a desarrollar un tumor mediastinal.2,3 A continuación, presentamos un paciente con historia familiar de NF tipo I, pero sin diagnóstico previo de esta afección al momento del examen.
PRESENTACIÓN DE CASO
DATOS CLÍNICOS RELEVANTES
Paciente masculino, de 19 años de edad; presenta asma bronquial como antecedente patológico y su antecedente patológico familiar es su padre con la enfermedad de Von Recklinghausen. El motivo de ingreso fue un dolor torácico referido como paravertebral izquierdo de 3 meses de evolución. Presenta piel con manchas color café con leche en región anterior del tórax y tetilla izquierda de más de 5 años de evolución (Fig. 1.).

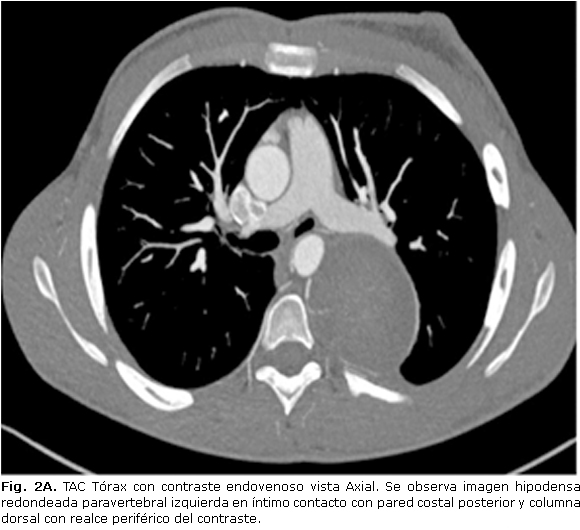
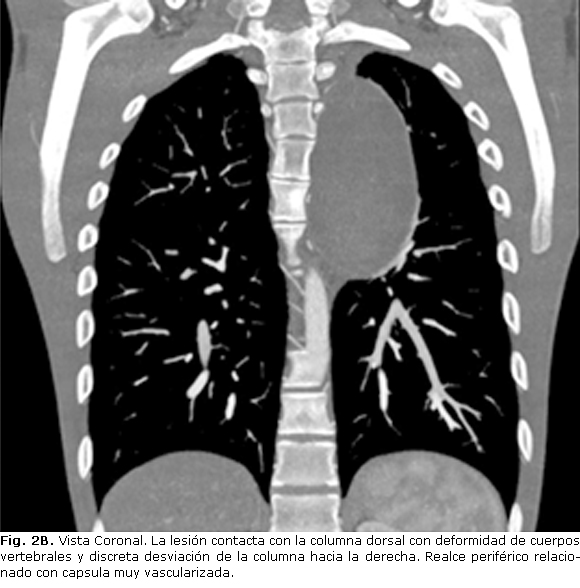
Ginecomastia izquierda. Cifosis dorsal y retraso mental discreto. Tórax: murmullo vesicular y vibraciones vocales disminuidas en hemitórax izquierdo. Se realizan estudios de imagen que se muestran a continuación donde se informa tumor mediastinal posterior (Fig. 2ª, 2B).
RESULTADOS
Se confirmó la presencia de un tumor sólido en mediastino posterior, asociado a NF Tipo I. Se conoció, además, que en muchos de estos enfermos la progresión hacia la malignidad ocurre hasta en un 34 %. El caso se discutió en el grupo multidisciplinario y se decidió tratamiento quirúrgico. La cirugía se realizó mediante toracotomía posterolateral izquierda. Se encontró un tumor de 11 cm en eje mayor, fijo a la región paravertebral izquierda con múltiples vasos nutricios. La lesión se resecó en su totalidad sin accidentes (Fig. 3A, B, C).

La evolución posquirúrgica fue satisfactoria. El diagnóstico histológico definitivo es neurofibroma.
Tres años después de la cirugía, el paciente se encuentra con buen estado general y se le ha diagnosticado su primer neurofibroma periférico en la región de la fosa poplítea. Hasta ese momento, no presentaba lesiones periféricas.
DISCUSIÓN
La NF tipo I se denominó inicialmente neurofibromatosis periférica debido a que algunos de los síntomas como manchas en la piel y tumores, parecían estar limitados al sistema nervioso periférico. Este nombre no es correcto, debido a que también pueden ocurrir tumores del sistema nervioso central. Este crecimiento está provocado por la mutación en un gen "supresor" de crecimiento tumoral en el 17q11.2 que codifica la neurofibromina: proteína que actúa como un supresor tumoral en condiciones normales que regula otra proteína celular que estimula el crecimiento y proliferación celular.4 La lesión característica -el neurofibroma- se origina a partir de las células de Schwann y fibroblastos de las vainas de los nervios periféricos.
Se inicia en la infancia y en la mayor parte de los enfermos el cuadro clínico es completo para la edad de 5 años, recrudeciéndose durante la pubertad, el embarazo y la menopausia. 5 Este es caso del paciente que presentamos, atendiendo a las primeras lesiones cutáneas observadas coincidiendo con el desarrollo puberal. Generalmente, se combinan manchas cutáneas y neurofibromas múltiples. En nuestro enfermo, es de notar la ausencia de lesiones tumorales periféricas que contrastan con la presencia de manchas cutáneas color "café con leche" en las localizaciones más habituales descritas tales como la axila y el tórax. Otras manifestaciones clínicas de la enfermedad y observadas en el enfermo fueron cifosis, retraso mental y ginecomastia. Todas ellas fueron descritas como parte del síndrome de NF tipo I. Lo anterior sumado al antecedente del padre que padece la enfermedad son criterios diagnósticos para confirmar la neurofibromatosis aun en ausencia de neurofibromas periféricos como se aclaró anteriormente.6
A pesar de la extrema variabilidad en la presentación y expresión fenotípica de esta enfermedad el desarrollo de tumores mediastinales no es un evento frecuente. Ocurre entre un 5-10 % de todos los pacientes, 3 por lo general se trata de un neurofibroma, originado de los nervios intercostales, simpático, frénico o vago. Las dos últimas localizaciones son más raras, y su ubicación sería en mediastino anterior o medio. Algo importante a tener en cuenta y que justifica la intervención quirúrgica, es la potencial transformación maligna que ocurre hasta en un 10 % de los enfermos. Esta cifra se eleva hasta un 34 % cuando se trata de tumores neurogénicos endotorácicos.7
El diagnóstico diferencial de un tumor mediastinal posterior en un paciente joven y en el contexto de NF tipo I siempre debe considerar en primer lugar el neurofibroma. Este representa el segundo tumor neurogénico más común constituyendo 10 % precedido solamente por el neurilemoma o schwannoma que incluye 40-50 % de todos los casos reportados.
En su conjunto, los tumores neurogénicos constituyen 20 % de todos los tumores mediastinales en el adulto.8 Un diagnóstico diferencial importante lo constituye el neurofibroma plexiforme, lesión infiltrativa local que se extiende al mediastino y de pronóstico diferente. Se ha reportado un incremento del riesgo para el desarrollo de otros tumores en estos enfermos, de histología variable como tumores de páncreas, colorectales y pulmón. Además, desde hace décadas se conoce que ciertos síndromes genéticos como el que describimos, tiene un riesgo aumentado de tumores mesenquimales o sarcomas, dentro y fuera del tórax, relacionado quizás con un incremento en la secreción de factor angiogénico.9-10 Lo singular y diferente en este paciente es la ausencia de neurofibromas periféricos, por lo que el mediastinal fue el primero en identificarse. Es decir, describimos un paciente con neurofibromatosis establecida y desarrollada, pero sin neurofibromas hasta ese momento.
Una explicación racional y posible a esta singularidad puede estar relacionado con el hecho de que el paciente pueda tener una variante muy infrecuente y poco reportada de NF tipo I llamada neurofibromatosis segmentaria que afecta a 1:36,000-40,000 individuos.11 Las lesiones se localizan solo en un dermatoma del cuerpo, de ahí la denominación de segmentaria. Sin embargo, sin un estudio genético confirmatorio no queda más que especular sobre este raro trastorno.
En cuanto a los estudios imaginológicos, sin bien la radiografía de tórax constituye el punto de partida para el diagnóstico inicial, se debe complementar con una tomografía axial computarizada (TAC) y resonancia magnética nuclear (RMN). En la TAC, los neurofibromas se describen como lesiones de baja densidad, con valores entre 15-20. La resonancia magnética nuclear es particularmente valiosa cuando se sospecha compresión de una raíz nerviosa o extensión intramedular. En nuestro paciente por la localización paravertebral izquierda de la lesión en íntimo contacto con la columna asociado con el dolor referido por el enfermo, se pensó esta posibilidad, por lo que el estudio fue realizado descartándose prolongación intramedular. La resección quirúrgica de la lesión es la única opción curativa para estos enfermos. El abordaje abierto o por toracotomía es el más comúnmente empleado, sin embargo, un número mayor de reportes se pueden encontrar hoy en día en que la resección se realiza por videotoracoscopia de manera exitosa.12
Conflicto de intereses
Los autores declaran que no hay conflicto de intereses.
REFERENCIAS BIBLIOGRÁFICAS
1. Gómez M, Batista O. Molecular diagnosis as a strategy for differential diagnosis and at early ages of neurofibromatosis type 1. Rev Med Chil 2015;143(10):1320-30.
2. Poyhonen M, Kytölä S, Leisti J. Epidemiology of neurofibromatosis type 1 (NF1) in northern Finland. J Med Genet. 2000;37(8):632-6.
3. Valeyrie-Allanore L, Ismaïli N, Bastuji-Garin S. Symptoms associated with malignancy of peripheral nerve sheath tumours: a retrospective study of 69 patients with neurofibromatosis 1. Br J Dermatol. 2005;153(1):79-82.
4. Abramowicz A, Gos M. Neurofibromin in neurofibromatosis type 1 - mutations in NF1gene as a cause of disease. Dev Period Med. 2014;18(3):297-306.
5. Petrák B, Bendová Š, Lisý J, Kraus J, Zatrapa T, Glombová M, et at. Neurofibromatosis von Recklinghausen type 1 (NF1) - clinical picture and molecular-genetics diagnostic. CeskPatol. 2015;51(1):34-40.
6. National Institutes of Health (NHI) Consensus Development Conference Statement: Neurofibromatosis. Bethesda, MD., USA July 13-15, 1987. Neurofibromatosis. 1988;1(3):172-8.
7. Krol EM, El-Fanek H, Borruso J. Solitary Neurofibroma with Malignant Transformation: Case Report and Review of Literature. Conn Med. 2015;79(4):217-9.
8. Ratbi MB, El Oueriachi F, Arsalane A, El Hammoumi MM, Kabiri el H. Surgery of benign neurogenic tumors in adults: single institution experience. Pan Afr Med J. 2014;19:288.
9. Sorensen SA, Mulvihill JJ, Nielsen A. Long-term follow-up of von Recklinghausen neurofibromatosis.Survival and malignant neoplasms. N Engl J Med. 1986;314:1010-5.
10. Gesundheit B, Parkin P, Greenberg M, Baruchel S, Senger C, Kapelushnik J, et at. The role of angiogenesis in the transformation of plexiform neurofibroma into malignant peripheral nerve sheath tumors in children with neurofibromatosis type 1.J Pediatr Hematol Oncol. 2010;32(7):548-53.
11. García-Romero M, Parkin P, Lara-Corrales I. Mosaic Neurofibromatosis Type 1: A Systematic Review. Pediatric Dermatology. 2015:1-9.
12. Lochowski MP, Brzeziński D, Kozak J. Videothoracoscopy in the treatment of benign neurogenic tumours of the posterior mediastinum. WideochirInne Tech Maloinwazyjne. 2014; 9(3):315-8.
Recibido: 29 de marzo de 2016.
Aprobado: 30 de abril de 2016.
Radamés Isaac Adefna Pérez. Hospital Clínico Quirúrgico Docente "Miguel Enríquez". La Habana, Cuba.
Correo electrónico: radamesiap@infomed.sld.cu


{kind=link}
{kind=link}