










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Farmacia
versión On-line ISSN 1561-2988
Rev Cubana Farm v.31 n.2 Ciudad de la Habana ene.-ago. 1997
Artículos Originales
Empresa Laboratorio Farmacéutico "Dr. Mario Muñoz"Esteroides c-funcionalizados. III. Participación del grupo 12b -benzoxi en la reacción de apertura de 9a, 11a-epoxiandrostanos
Fernando Verdecia Navarro,1 Daniel Calcines Huerta,2 Carlos Pérez Martínez3 y Pedro Ortiz del Toro32 Investigador Titular. Centro de Investigación y Desarrollo de Medicamentos.
3 Profesor Titular. Facultad de Química. Universidad de La Habana.
RESUMEN
Se describió la síntesis de la 5a-androstan-3b ,9a ,11b ,12b -tetrol-17-ona y su acetónido por tratamiento del 9a ,11a-epoxi-12b-benzoxiderivado correspondiente con HClO4 al 70 %. Las estructuras de los productos obtenidos se asignaron empleando RMN-1H y 13C.Descriptores DeCs: ANDROSTANOS/síntesis química; ANALISIS ESPECTRAL.
La síntesis de polihidroxiandrostanos vía apertura de 12b-acetoxi-9a , 11a-epoxiderivados con HClO4 70 %,1 ocurre con bajo rendimiento. Esto se debe a que, conjuntamente con la apertura del anillo oxiránico, en cuya reacción participa el grupo acetoxilo, se observa la hidrólisis de éste.
En un intento por mejorar el rendimiento de esta reacción, decidimos estudiar la posibilidad de utilizar el grupo benzoxi en lugar del acetoxi, atendiendo a que el primero debe formar un carbocatión más estable y no se debe hidrolizar con tanta facilidad en medio alcalino. Reacciones de este tipo han sido estudiadas por varios autores.2 De corroborarse esto, el método tendría un interés preparativo.
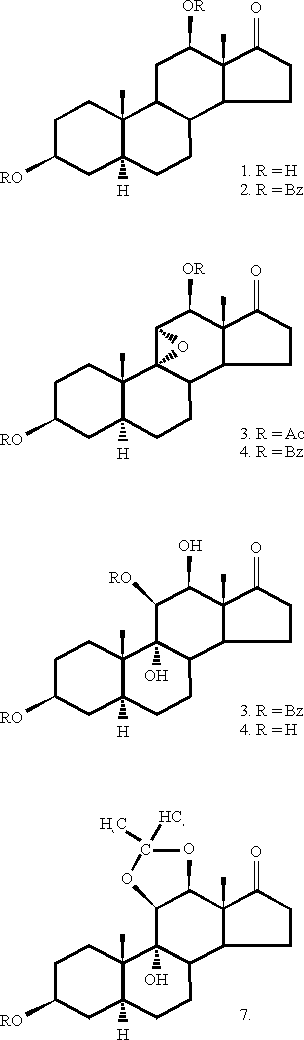
El epóxido de partida 4 (figura 1) se obtuvo por benzoilación de 1 (cuya síntesis ha sido reportada anteriormente3) y ulterior epoxidación (ácido metacloroperbenzoico) del 9(11)-dehidroderivado resultante.2
MÉTODOS
Los puntos de fusión se determinaron en capilar abierto y no están corregidos. Los espectros IR (u ,cm-1) se registraron en un espectrómetro IR-75 Carl Zeiss en pastillas de KBr.Los espectros de RMN -1H y -13C (d, p.p.m.) se registraron en un equipo Bruker ACF-90 (resonancia protónica a 250,13 MHz y 13Ca 62,9 MHz) utilizando como solvente CDCl3 (para los compuestos 6 y 7 se utilizó una mezcla CDCl3/CD3OD); los desplazamientos químicos se reportaron con respecto al TMS como referencia interna. Se utilizó la técnica de edición DEPT para determinar el número de protones unidos directamente a cada átomo de carbono. Los ángulos de rotación se determinaron en un polarímetro Polartronic D. La cromatografía en capa delgada (CCD) se realizó sobre cromatoplacas de silicagel 60F254 de 0,25 mm de espesor.
5a -andros-9(11)-en-3b ,12b -diol-17-ona dibenzoato(2)
A una solución de 1,0 g (3,29 mmol) del compuesto 1 en 5 mL de piridina se adicionó, a temperatura de 0,5 oC y con agitación, 1,15 mL de cloruro de benzoílo (9,9 mmol). La mezcla de reacción se agitó 0,5 h a 5 oC y después 1 h a temperatura ambiente. Se vertió sobre 100 mL de solución de NaHCO3 al 2 % y se agitó 0,5 h. El precipitado se filtró, se lavó con agua a 60-70 oC hasta desaparición del olor a piridina y se secó a 70 oC bajo vacío. El producto crudo se recristalizó de CH2Cl2-MeOH. Rend: 1,55 g (92 % del teórico). Tf: 198-201 oC, [a ]D-18 o (c=1, CHCl3). IR: 1 705 (ancha, C=O); 1 264 y 1 111 (éster). RMN-1H: 1,09 (6H, s, 18-CH3 y 19-CH3); 4,92 (H, m, 3-H); 5,24 (H, m, 11-H); 5,75 (H, m, 12-H); 7,5-8,0 (10H, m, fenilos). RMN-13C: carbonos metílicos: 9,2 (C-18); 17,8 (C-19); carbonos meti-lénicos: 21,9; 27,3; 27,8; 31,8; 33,9; 34,9; 36,1; carbonos metínicos: 36,0; 42,7; 47,5; carbonos cuaternarios: 38,1 (C-10); 48,6 (C-13); carbonos olefínicos: 150,8 (C-9); 117,6 (C-11); carbonos carbonílicos: 216,8 (C-17); 166,0 (grupos benzoílo); carbonos enlazados a oxígeno: 72,5 (C-3); 73,5 (C-12).
5a -androstan-9a , 11a -epoxi-3b , 12b-diol-17-ona dibenzoato(4)
A una solución de 1,5 g (2,93 mmol) del compuesto 2 en 50 mL de CHCl3 se adicionó a temperatura de 20-25 oC y con agitación, 2,38 g de ácido m-cloroperbenzoico (85 % de pureza; 11,7 mmol). La mezcla de reacción se agitó 1 h y después se dejó en reposo 72 h a temperatura ambiente. Se diluyó con 25 mL de CHCl3, se lavó con solución de Na2SO3 al 10 %, después con solución de NaHCO3 al 5 % y, finalmente, con agua hasta neutralidad. La solución orgánica se secó sobre Na2SO4 anhidro y el solvente se destiló hasta sequedad bajo vacío. Se obtuvo 1,55 g (100 % del teórico). El producto crudo se recristalizó de CH2Cl2-MeOH. Rend: 1,3 g (84 % del teórico). Tf: 252-255 oC; [a ]D 0o (c=1, CHCl3). IR: 1 705 (ancha, C=O); 1 267 y 1 107 (éster). RMN-1H: 1,09 (3H, s, 18-CH3); 1,14 (3H, s, 19-CH3); 3,0 (H, m, 11-H); 4,85 (H, m, 3-H); 5,23 (H, m, 12-H); 7,5-8,0 (10H, m, fenilos). RMN-13C: carbonos metílicos: 10,5 (C-18); 15,4 (C-19); carbonos metilénicos: 21,4; 26,7; 27,1; 28,2; 28,7; 33,5; 36,2; carbonos metínicos: 34,1; 39,7; 39,9; carbonos cuaternarios: 36,8 (C-10); 48,6 (C-13); carbonos carbonílicos: 217,6 (C-17); 166,0 (grupos benzoílo); carbonos enlazados a oxígeno: 57,8 (C-11); 68,8 (C-9); 70,8 (C-12); 73,2 (C-3).
5a -androstan-3b , 9a , 11b , 12b-tetrol-17-ona 3,11-dibenzoato(5)
A una solución de 500 mg (0,95 mmol) del compuesto 4 en 5 mL de CH2Cl2 y 5 mL de cetonitrilo se adicionó, a temperatura ambiente y con agitación, 0,8 mL de HClO4 70 %. Se agitó durante 2 h, se diluyó con agua y se separó la capa orgánica. La capa acuosa se extrajo con CH2Cl2 y los extractos orgánicos reunidos se lavaron con solución de NaHCO3 al 5 % y con agua hasta neutralidad. La solución orgánica se secó sobre Na2SO4 anhidro y el solvente orgánico se destiló hasta sequedad bajo vacío.
El residuo crudo (480 mg) se cromatografió a través de una columna de silicagel 60 (70-hexano-acetato de etilo 2:1 como eluyente). Se obtuvo 410 mg (79 % del teórico) del compuesto 5. Tf: 267-269 oC (CH2Cl2-MeOH); [a ]D +35 o (c=1, CHCl3). IR: 3 519 (OH); 1 705 (ancha, C=O); 1 269 y 1 113 (éster). RMN-1H: 1,17 (3H, s, 18-CH3); 1,05(3H, s, 19-CH3); 5,79 (H, d, 11-H); 4,90 (H, m, 3-H); 4,35 (H, dd, J=3,6/2,4 Hz, 12-H); 7,3-8,1 (10H, m, fenilos). RMN-13C: carbonos metílicos: 9,4 (C-18); 16,8 (C-19); carbonos metilénicos: 21,2; 24,2; 26,9; 27,4; 30,0; 33,4; 35,2; carbonos metínicos: 34,0; 36,6; 43,0; carbonos cuaternarios: 40,5 (C-10); 51,0 (C-13); carbonos carbonílicos: 219,8 (C-17); 165,6; 165,2 (grupos benzoílo); carbonos enlazados a oxígeno: 70,2 (C-12); 73,4 (C-3); 75,0 (C-11); 78,0 (C-9).
5a -androstan-3b , 9a , 11b , 12b -tetrol-17-ona(6)
Una mezcla de 410 mg (0,75 mmol) del compuesto 5, 15 mL de MeOH y 3 mL de solución al 10 % de NaOH se reflujó durante 2 h. La mezcla de reacción se concentró, se diluyó con 20 mL de agua y se extrajo con acetato de etilo. La solución orgánica se lavó con agua hasta neutralidad, se secó sobre Na2SO4 anhidro y el solvente se destiló hasta sequedad bajo vacío. El residuo crudo (252 mg) se cromatografió a través de una columna de silicagel 60 (70-230 mesh) y n-hexano-acetato de etilo 1:3 como eluyente. Se obtuvieron 210 mg (83 % del teórico) del compuesto 6. Tf: 237-240 oC (MeOH-CHCl3); [a ]D +38o (c=0,5; MeOH). IR: 3 500-3 300 (ancha, OH); 1 714 (C=O). RMN-1H: 1,09 (3H, s, 18-CH3); 1,17 (3H, s, 19-CH3); 3,50 (H, m, 3-H); 3,83 (H, d, J=3,6 Hz, 12-H); 3,92 (H, d, 11-H). RMN-13C: carbonos metílicos: 9,8 (C-18); 16,4 (C-19); carbonos metilénicos: 21,9; 24,9; 28,4; 0,5; 31,3; 36,2; 37,9; carbonos metínicos: 33,7; 37,1; 44,1; carbonos cuaternarios: 41,5 (C-10); 52,4 (C-13); carbono carbonílico: 222,2 (C-17); carbonos enlazados a oxígeno: 1,0; 71,1 (C-3 y C-12); 76,6 (C-11); 78,6 (C-9).
5a -androstan-3b , 9a , 11b , 12b -tetrol-11,12-isopropilidendioxi-17-ona(7)
A una solución de 190 mg (0,56 mmol) de 6 en 30 mL de acetona se adicionó 0,27 mL de HClO4 al 70 % y se agitó 30 min a temperatura ambiente y después se dejó en reposo durante 3 h. La mezcla de reacción se neutralizó con solución de NaHCO3 al 10 %, se evaporó el solvente, se diluyó con agua y se extrajo con acetato de etilo. La solución se secó sobre Na2SO4 anhidro y el solvente se destiló hasta sequedad bajo vacío. Se obtuvo 0,21 g (99 % del teórico) del compuesto 7, el cual se purificó por recristalización de acetato de etilo. Tf: 221-224 oC. IR: 3 556 (OH); 1 722 (C=O). RMN-1H: 1,01(3H, s, 18-CH3); 1,12 (3H, s, 19-CH3); 1,36 y 1,52 (6H, s, isopropilideno); 3,60 (H, m, 3-H); 4,12 (H, d, 12-H); 4,20 (H, d, 11-H). RMN-13C: carbonos metílicos: 12,1 (C-18); 14,1 (C-19); 25,0 y 25,6 (isopropilideno); carbonos metilénicos: 21,4; 23,9; 27,2; 29,2; 30,8; 35,2; 37,3; carbonos metínicos: 34,2; 36,6; 40,4; carbonos cuaternarios: 40,8 (C-10); 51,7 (C-13); carbonos carbonílicos: 219,7 (C-17); carbonos enlazados a oxígeno: 70,6 (C-3); 75,6 (C-11); 77,3 (C-9); 80,3 (C-12); 108,8 (isopropilideno).
RESULTADOS
El compuesto 4, a diferencia de su análogo diacetilado, no es soluble en metanol; por esta razón, la reacción de apertura se realizó en una mezcla de partes iguales de cloruro de metileno y acetonitrilo. En estas condiciones, 3 ni se hidroliza ni sufre apertura del epóxido.El tratamiento del epóxido 4 con HClO4 70 % en una mezcla de partes iguales de cloruro de metileno y acetonitrilo dio el producto de apertura 5 con el 79 % de rendimiento, muy superior al 25 % obtenido en el caso de los 9a , 11a-epoxi-12b-acetoxiandrostanos.
El compuesto 5 en HClO4 70 %-Me2CO durante 24 h no forma el acetónido (11-OBz); sin embargo, si se le refluja durante 1 h en Ac2O puede observarse por CCD la formación de un producto menos polar, que sólo puede deberse a la acetilación del OH ecuatorial en C-12.
El compuesto 6, obtenido por hidrólisis de 5, sí forma el acetónido 7 al ser tratado en iguales condiciones. La estructura de este último compuesto fue ratificada mediante RMN-1H y -13C.
DISCUSIÓN
La participación del grupo 12b -acetoxi en la reacción de apertura de 9a ,11a -epoxiandrostanos ha sido establecida con anterioridad.4 En ese trabajo postulamos que ocurría una apertura cis del anillo oxiránico con la participación del grupo acetoxi vecinal, por cuanto en las condiciones estudiadas el epóxido aislado no reacciona.Los espectros de RMN-1H y -13C demostraron que, contrariamente a lo que suponíamos, la dirección de apertura del epóxido es trans-diaxial, observándose una migración 12?11 del grupo benzoílo, lo que indica que más que una apertura del anillo oxiránico con el H2O actuando como nucleófilo externo (A, figura 2), lo que parece ocurrir es una hidratación del ion cíclico intermedio (B).
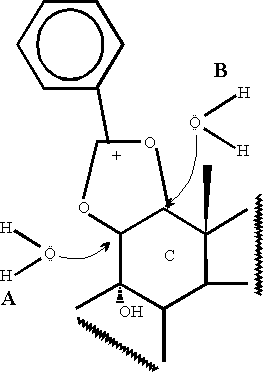
La configuración del producto de apertura 5 se estableció sobre la base de los espectros de RMN-13C y -1H. El metilo C-18 aparece a d =9,4 p.p.m. con un apreciable blindaje con respecto al carbono equivalente en la 5b -andros-tan-17-ona (13,8 p.p.m.5), lo que indica que el sustituyente ocupa la posición 12b . La constante de acoplamiento interprotónica vecinal 11-12 de 3,7 Hz corresponde a una configuración ecuatorial para el hidrógeno en 11 (H11 ecuatorial-H12 axial. El sustituyente en 12b es un grupo hidroxilo que forma enlace de hidrógeno intramolecular con el grupo carbonilo en 17. El lento intercambio de este protón hidroxílico permite observar el acoplamiento vecinal de éste con el H12a (J=2,4 Hz) y una asignación inequívoca de todas las señales.
SUMMARY
The synthesis of 5 a -andostrane- 3b , 9a , 11b , 12b tetrol-17-ona and its aceto-nid is described by the treatment of the corresponding 9a , 11a , epoxyl-12b-benzo derivative with HClO4 70 %. The structures of the products obtained were assigned using NMR-1H and 13C.Subject headings: ANDROSTANES/chemical; SYNTHESIS; SPECTRUM ANALYSIS.
REFERENCIAS BIBLIOGRÁFICAS
- Verdecia Navarro F, Calcines Huerta D, Merás Cepero M. Esteroides C-funcionalizados. II . Participación del grupo 12b -acetoxilo en la reacción de apertura de 9a , 11a -epoxiandrostanos. Rev Cubana Farm 1993;27(2):75-81.
- 2.Koizumi N, Ikekawa N, Benzoyl Group participation in epoxide ring opening of 26-benzoxy-24,25- -epoxicholesterol derivatives. Reinvestigation of the stereochemistry. Chem Pharm Bull 1983;31(10):3465--70.
- 3.Verdecia Navarro F, Calcines Huerta D, Merás Cepero M. Esteroides C-funcionalizados. I. Intermediarios de síntesis. Rev Cubana Farm 1993;27(2):81-7.
- 4.Verdecia Navarro F, Calcines Huerta D, Vélez Castro H. Acerca de la reactividad de 9a , 11a -epóxidos de la tigogenina con ácido perclórico y yoduro de metilmagnesio. Rev Cubana Farm 1989;23(1-2):41-7.
- 5.Blunt JW, Stothers JB. 13C RMN Spectra of steroid. Org Magn Resonance 1977;9(8):439.
Lic. Fernando Verdecia Navarro . Empresa Laboratorio Farmacéutico "Dr. Mario Muñoz". Hacendados No. 1, municipio Habana Vieja, La Habana, Cuba.

