










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Farmacia
versión On-line ISSN 1561-2988
Rev Cubana Farm v.34 n.2 Ciudad de la Habana Mayo-ago. 2000
Centro de Investigación y Desarrollo de Medicamentos
Validación del método analítico para la determinación de 3 vitaminas hidrosolubles en un suplemento vitamínico
Iverlis Díaz Polanco1 y Ofelia Fariñas Suárez2RESUMEN
Se presentan los resultados obtenidos en la validación de un método analítico por cromatografía líquida de alta resolución, para la determinación de tiamina mononitrato, piridoxina clorhidrato y nicotinamida en el suplemento nutricional neovitamin II, el cual se diseñó para separar las vitaminas entre sí, con la utilización de una columna RP-18 de 25 cm y un detector UV-Visible. Dicho método se empleó para el control de la calidad y la estabilidad de este producto. El método fue validado siguiendo una metodología de trabajo elaborada previamente en un Protocolo de Validación, donde se analizaron diferentes parámetros como son: linealidad, exactitud, precisión, selectividad, límites de detección y cuantificación, adecuación del sistema y estabilidad de las soluciones. Se obtuvieron resultados satisfactorios y se comprobó de esta forma la validez del método analítico.Descriptores DeCS: SUPLEMENTOS DIETETICOS/análisis; TIAMINA/análisis; PIRIDOXINA/análisis; NIACINAMIDA/análisis; CALIDAD DE LOS MEDI-CAMENTOS; ESTABILIDAD DE MEDICAMENTOS; CROMATOGRAFIA LIQUIDA DE ALTA PRESION/métodos.
El neovitamin II es un suplemento multivitamínico esencial como requerimiento nutricional para un crecimiento normal del organismo humano, es administrado fundamentalmente a embarazadas, ancianos y pacientes que presenten estados hipovitamínicos. Está constituido por tiamina mononitrato (3,85 mg), nicotinamida (22 mg), piridoxina clorhidrato (2,5 mg), riboflavina (1,84 mg), ácido fólico (0,325 mg), vitamina A palmitato (32,5 mg) y cianocobalamina (0,072 mg).
Una de las grandes dificultades en el desarrollo de técnicas analíticas para complejos vitamínicos es contar con métodos específicos, principalmente si los métodos a utilizar se emplearan en estudios de estabilidad.
Aparecen por primera vez reportadas en la Farmacopea de los Estados Unidos Edición 23, técnicas oficiales para suplementos nutricionales, de manera que se determinan las vitaminas por cromatografía líquida de alta resolución, simultáneamente con el resto de las vitaminas hidrosolubles.2
El objetivo del presente trabajo es demostrar la validez del método analítico desarrollado en el Departamento de Productos Naturales del Centro de Investigaciones y Desarrollo de Medica-mentos (CIDEM) para la determinación de las vitaminas hidrosolubles: tiamina mononitrato, piridoxina clorhidrato y nicotinamida presentes en el neovitamin II. Para ello se seleccionaron una serie de parámetros a determinar, como son: linealidad, exactitud, precisión, selectividad, límites de detección y cuantificación, adecuación del sistema y estabilidad de las soluciones.
MÉTODOS
Todas las determinaciones se realizaron en un cromatógrafo líquido de alta resolución equipado con una columna de fase reversa LiChrosorb RP-18, de 25 cm de longitud, un detector UV-Vis a una longitud de onda de 280 nm, un flujo de alrededor de 1 mL/min y un loop de 20 µL. Entre los reactivos y soluciones utilizados se encuentran agua purificada, buffer de hidrógeno fosfato de potasio 0,03 mol/L y pH 2,7 la fase móvil buffer-metanol (99-1) y las soluciones de referencia de cada vitamina:
- Estándar de tiamina mononitrato: se pesó con exactitud 38,5 mg de tiamina mononitrato, se transfirió cuantitativamente a un matraz aforado de 100 mL, se añadió 70 mL de agua, se agitó hasta completar disolución y se llevó a volumen con el mismo solvente (solución T).
- Estándar de piridoxina clorhidrato: se pesó con exactitud 25,0 mg de piridoxina clohidrato, se transfirió cuantitativamente a un matraz aforado de 100 mL, se añadió 70 mL de agua, se agitó hasta completar disolución y se llevó a volumen con el mismo solvente (solución P).
- Estándar de nicotinamida: se pesó con exactitud 220,0 mg de nicotinamida, se transfirió cuantitativamente a un matraz aforado de 100 mL, se añadió 70 mL de agua, se agitó hasta completar disolución y se llevó a volumen con el mismo solvente (solución N).
- Solución E: se tomaron alícuotas de 5,0 mL de cada una de las soluciones T, P y N, se trasvasaron cuantitativamente a un matraz aforado de 25 mL y se llevó a volumen con agua.
Métodos analíticos para la determinación de los parámetros a estudiar
Los parámetros a estudiar se seleccionaron en función de las características y de los objetivos del método analítico utilizado y el rango de concentraciones en que se encuentra cada vitamina en la formulación. La metodología aplicada al análisis de cada parámetro fue descrita por Mestony5 (Analytical Methods Validation,1991), Loftus Bernard T3 y Castro CM.4 Linealidad. Se analizó sobre soluciones de referencia a las concentraciones de 50, 80, 100, 120 y 150 % de la cantidad declarada para cada vitamina. El análisis se realizó por duplicado. De las soluciones de referencia de cada vitamina se tomaron alícuotas, se transfirieron cuantitativamente a un matraz aforado y se llevó a volumen con agua.
Las áreas obtenidas para cada concentración se procesaron según el método estadístico MICROSTA; se determinó el coeficiente de correlación y se aplicaron las pruebas de linealidad y proporcionalidad.
Exactitud. Se prepararon 5 muestras por duplicado a las concentraciones de 50, 80, 100, 120 y 150 % de la cantidad declarada para cada vitamina. Para la preparación de las muestras se pesó con exactitud alrededor de 127,0 mg de polvo de placebo equivalente a una tableta, se transfirió cuantitativamente a un matraz aforado de 50 mL, se añadió 20 mL de agua, se agitó durante 10 min y se adicionaron alícuotas de las soluciones T, P y N hasta obtener las concentraciones en estudio. Se llevó a volumen con el mismo solvente y se filtró por papel de filtración lenta, desechando los primeros mililitros del filtrado.
Se determinó la recuperación media o el porcentaje de recobro y la desviación estándar relativa (DER) entre las concentraciones obtenidas para concentración teórica estudiada.
Precisión. Este parámetro se determinó mediante los ensayos de repetibilidad y reproducibilidad.
Para el ensayo de repetibilidad se seleccionó un lote industrial, con el cual se procedió de la forma siguiente: se prepararon 6 muestras a una concentración del 100 % de la cantidad declarada para cada vitamina y se analizó ese día por el mismo analista.
En ensayo de reproducibilidad se analizó con el mismo lote utilizado para la repetibilidad y se prepararon 3 muestras a la concentración del 100 % de la cantidad declarada para cada vitamina, las que se analizaron en 2 d diferentes.
Preparación de las muestras. Se trituraron no menos de 20 tabletas y se pesó con exactitud el equivalente a la masa promedio, se transfirió cuantitativamente a un matraz aforado de 50 mL, se añadió 30 mL de agua, se agitó durante 10 min, se llevó a volumen con el mismo solvente y se filtró por papel de filtración lenta, desechando los primeros mililitros del filtrado. En ambos ensayos se determinó la DER entre las concentraciones obtenidas en los 2 d de análisis.
Selectividad. El estudio de este parámetro se demostró mediante 3 muestras placebos, realizando comparaciones entre las soluciones de referencia antes y después de sometidas a un proceso de degradación. Para esto las soluciones de referencia se analizaron recién preparadas y posteriormente se almacenaron en frascos de cristal ámbar a 70 °C durante 6 d; al finalizar este tiempo se analizaron nuevamente por triplicado. Las muestras se prepararon como se indica en el parámetro de exactitud sin la adición de las alícuotas de las soluciones de referencia.
Límites de detección y cuantificación. Se realizó posteriormente a la determinación del parámetro linealidad, donde se aplicó una calibración lineal del tipo y = bx + a. Se utilizó para determinar dichos parámetros la medición de 3 soluciones de referencia de las vitaminas en estudio, tomando como patrón la concentración del 50 % y 2 con-centraciones por debajo de ésta, 20 y 35 %. El análisis se realizó por triplicado para cada concentración.
Los resultados obtenidos se evaluaron estadísticamente calculando la media y la DER de las concentraciones; además se calculó la recta de regresión tomando como Y la respuesta y como X las concentraciones. Se definió la ecuación y se determinó la respuesta a concentración cero por extrapolación al origen de la recta calculada. El valor de Y será Ybl. Se determinó la desviación estándar de las respuestas a concentración cero por extrapolación al origen de la recta calculada, tomando como Y la DER de las respuestas y como X las concentraciones. El valor de Y será Sbl. Se calcularon los límites de detección y cuantificación mediante las fórmulas siguientes:
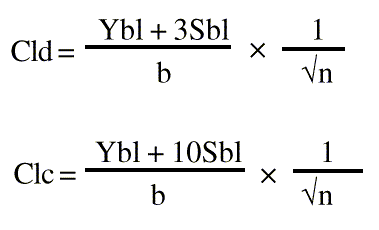
donde:
Cld: límite de detección (µg/mL)
Clc: límite de cuantificación (µg/mL)
b: valor de la pendiente de la recta de calibración
n: número de muestras
Adecuación del sistema. Este parámetro se analizó con 6 muestras de un lote industrial a la concentración del 100 % de la cantidad declarada para cada vitamina. La preparación de las muestras y las soluciones de referencia se realizó como se indica en el parámetro de exactitud. En los cromatogramas obtenidos se determinó el factor de cola (T), la resolución (R) y la precisión de la inyección (Cv). Los cálculos se realizaron con las fórmulas siguientes:
T = W.05/2f
donde:
W . 05: ancho del pico al 5 % de la altura
f: distancia de la orilla principal al pico máximo
R = 2t2 - t1 / W2 + W1
donde:
t 2 y t1: tiempos de retención
W2 y W1: ancho de las porciones extrapoladas
Cv = S/![]() (100)
(100)
donde:
S: desviación estándar absoluta
X: media
Estabilidad de las soluciones. Debido a que las vitaminas se degradan con gran facilidad, se determinó este parámetro. Para ello se midieron 3 soluciones de referencia de cada vitamina a las 3, 6 y 9 h de su preparación, almacenadas en matraces aforados protegidos de la luz.
En el análisis de cada parámetro se calculó la concentración de las vitaminas, expresada en porcentaje, según la fórmula siguiente:
C (%) = Cm/Ct (100)
donde:
Cm: valor promedio de la concentración de tiamina mononitrato, nicotinamida y piridoxina clorhidrato, según corresponda en microgramos por mililitro.
Ct: valor promedio de la concentración teórica de tiamina mononitrato, nicotinamida y piridoxina clorhidrato, según corresponda en microgramos por mililitro.
RESULTADOS
Como resultado del análisis de la linealidad del método analítico se obtuvieron valores de coeficiente de correlación r menor o igual a 0,99, intercepto de la recta de regresión a cero y DER menor o igual al 5 %. Los resultados del análisis del parámetro de exactitud se muestran en la tabla 1.
| Concentraciones | | | | |||
| % . | | | | | | |
| 50 | | | | | | |
| 80 | | | | | | |
| 100 | | | | | | |
| 120 | | | | | | |
| 150 | | | | | | |
El análisis de los ensayos de repetibilidad y reproducibilidad se presentan en las tablas 2 y 3 con valores de DER menores que los criterios de aceptación establecidos (menor o igual que 2,0 % y menor e igual que 3,0 %, respectivamente).
| Vitaminas | | |
| Tiamina mononitrato | ||
| (70,0 µg/mL) | | |
| Piridoxina clorhidrato | ||
| (45,0 µg/mL) | | |
| Nicotinamida | ||
| (440,0 µg/mL) | | |
| | ||
| | ||
| Vitaminas | | |
| Tiamina mononitrato | | |
| DER | | |
| Piridoxina clorhidrato | | |
| DER | | |
| Nicotinamida | | |
| DER | | |
En el estudio del parámetro de selectividad se observó que las vitaminas en solución disminuyen su concentración cuando son sometidas a un proceso de degradación térmica. En el caso de la tiamina mononitrato se parte de una concentración inicial de 103,6 % y se obtiene una concentración final de 86,0; la piridoxina clohidrato varía su concentración desde 101,0 hasta 90,5 % y la nicotinamida de 104,0 a 103,0 %. Además los excipientes de la formulación no absorben a la misma longitud de onda de los principios activos.
Los límites de detección y cuanti-ficación obtenidos fueron los siguientes:
- tiamina mononitrato: Cld = 0,0055 µg/mL y Clc = 0,0475 µg/mL
- piridoxina clohidrato : Cld = 0,059 µg/mL y Clc = 0,1436 µg/mL
- nicotinamida: Cld = 0,926 µg/mL y Clc= 2,97 µg/mL
| Vitaminas | | | |
| Tiamina mononitrato | | | |
| Piridoxina clorhidrato | | | |
| Nicotinamida | | |
Todas las vitaminas se mantienen estables en solución hasta las 9 h después de su preparación, excepto la tiamina mononitrato que ya a las 6 h se encuentra en menos del 90 %.
DISCUSIÓN
Como resultado del análisis de la linealidad del método, dicho parámetro cumplió con los criterios de aceptación establecidos, y hubo una relación lineal entre las concentraciones de las vitaminas estudiadas y las áreas obtenidas para cada una. Como se observa en la tabla 1, se obtuvieron valores de desviación estándar relativa y porcentaje de recobro (% rec) dentro de los límites establecidos en el protocolo de validación (DER menor o igual que 2,0 % y % rec = 97,0 - 103,0 %, y hubo una relación lineal entre los valores de referencia, por lo que se considera que el método es exacto.
En el parámetro de repetibilidad, 6 determinaciones de cada vitamina fueron necesarias para asegurarse que el método es preciso, con valores de desviación estándar relativa menores que 2,0 %. Según los resultados obtenidos en la reproducibilidad se puede concluir que el método es reproducible cambiando el día de análisis, ya que en ambos días se obtuvieron valores de coeficientes de variabilidad menores que 2,0 %.
El método cromatográfico es selectivo y diferencia los principios activos de sus productos de degradación, además los excipientes de la formulación no interfieren en la determinación de las vitaminas.
El método analítico cumple con el parámetro de límites de detección y cuantificación, ya que permite detectar y cuantificar concentraciones muy bajas de las 3 vitaminas presentes en el neovitamin II.
El sistema cromatográfico empleado es adecuado para la determinación de dichas vitaminas hidrosolubles en el neovitamin II, pues las variables analizadas en el parámetro de adecuabilidad cumplieron con los límites establecidos (T = 0,85-1,15; R>1,5 y Cv menor o igual que 2,0 %), o sea, se obtienen picos finos, bien definidos y con buena resolución entre ellos.
Protegiendo de la luz las soluciones de cada vitamina se logra una adecuada estabilidad de éstas, ya que conservan las características físico-químicas y sus concentraciones iniciales no varían considerablemente.
Teniendo en cuenta los resultados obtenidos, la técnica analítica propuesta para el control de la calidad y estudio de estabilidad del neovitamin II, es válida para obtener resultados satisfactorios en las 3 vitaminas estudiadas; así se demuestra en los parámetros de linealidad, precisión exactitud, límites de detección, cuantificación y especificidad. Como método cromatográfico es adecuado y las soluciones son estables de forma que garantizan su integridad durante un largo período.
SUMMARY
The results obtained in the validation of an anlytical method by high resolution liquid chromatography for the determination of thiamine mononitrate, pyridoxine hydrochloride and nicotinamide in the Neovitamin dietary supplement are presented. It was designed to separate vitamins among themselves, using a RP-18 column of 25 cm and a UV Visible detector. This method was used for controlling the quality and stability of the product. This method was validated according to a working methodology that was previously prepared in a Validation Protocol where paramaters such as lineality, accuracy, precision, selectivity, limits of detection and quantitation, system adequacy and the solution stability were analyzed. The results were satisfactory and the validity of the analytical method was proved. Subject headings: DIETARY SUPPLEMENTS/analysis; THIAMINE/analysis; STABILITY; CHROMATOGRAPHY, HIGH PRESSURE LIQUID/methods.
REFERENCIAS BIBLIOGRÁFICAS
- Drug Information for the Health Care Professional USP DI 15 ed. United States Pharmacopeial Convention, Inc. Rockville: Mack Printing; 1995:2802.
- United States Pharmacopeia USP 23. United States Pharmacopeial Convention, Inc. Rockville: Mack Printing; 1995:2143-6,2162-8.
- Pasteelnick LA: Analtycal Methods Validation. En: Loftus Bernard T, Nash RA, eds Pharmaceutical process validation. New York, Basel: Marcel Dekker; INC,1984:251-66.
- Castro CM, Gascón FS, Pujol FM, Sans RM, Pla VL. Validación de métodos analíticos. Asociación española de Farmacéuticos de la Industria (AFEI), Madrid. Comisión de Normas, Buena Fabricación y Control de la Calidad, 1989:8,38,43-44,57,61.
Recibido: 4 de diciembre de 1999. Aprobado: 21 de enero del 2000.
Lic. Iverlis Díaz Polanco. Centro de Investigación y Desarrollo de Medicamentos. Calle 19 de mayo No.13 esquina a Amézaga, municipio Plaza de la Revolución, Ciudad de La Habana, Cuba.

