










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Farmacia
versión impresa ISSN 0034-7515versión On-line ISSN 1561-2988
Rev Cubana Farm v.34 n.3 Ciudad de la Habana sep.-dic. 2000
Instituto de Medicina Tropical "Pedro Kourí"
Terapia antiviral para VIH-SIDA
Alicia Tarinas Reyes,1 Rolando David Tápanes Peraza2 y Lorenzo Jorge Pérez Ávila3RESUMEN
En los últimos años, muchos agentes antivirales nuevos han sido incorporados a la quimioterapéutica. En esta revisión se resumen tanto los fármacos establecidos de años atrás como los nuevos medicamentos desarrollados para el tratamiento de individuos infectados por VIH. El AZT fue el primero aprobado en marzo de 1987, le siguió el ddl (1991), ddC (1992), d4T (1994), 3TC (1995). Luego fue aprobado el primer inhibidor de proteasa, saquinavir en diciembre de 1995, seguido de ritonavir (1996), indinavir (1996), nelfinavir (1997); además de otros inhibidores de la reverso transcriptasa como nevirapine (1996), delavirdine (1997), efavirenz (1998), entre otros. En estos momentos se siguen buscando y desarrollando nuevas terapias alternativas para esta afección. En este trabajo se exponen algunas de las características de dichos medicamentos, como son: mecanismos de acción (sobre qué enzima actúa cada uno y cómo lo hacen, el ciclo viral), dosificación, incompatibilidades y reacciones adversas.Descriptores DeCS: AGENTES ANTIVIRALES/quimioterapia; SINDROME DE INMUNODEFICIENCIA ADQUIRIDA/farmacología; QUIMIOTERAPIA COMBINADA.
La aparición anualmente en la práctica médica de 2 ó 3 nuevos agentes antivirales desde 1995 ha permitido avances sin precedente en el tratamiento del VIH. La terapia combinada ha demostrado ser la más efectiva para tratar el VIH. De todo el potencial de la terapia combinada con drogas disponibles se ha realizado solo un número limitado de posibles combinaciones incorporando nuevas drogas que han sido totalmente probadas. La terapia combinada puede aumentar la supresión viral, prevenir la resistencia a los medicamentos, optimizar la exposición a la droga y simplificar la dosis, pero también puede resultar en un antagonismo farmacológico, concentraciones subterapéuticas de las drogas y toxicidad inesperada.1
Los antivirales se clasifican según su mecanismo de acción en:
- Inhibidores de la reverso transcriptasa (IRT):
- Nucleósidos (IRTNs):
- AZT (Retrovir, zidovudina)
- ddl (Videx, didanosina)
- ddC (Hivid, zalcitabina)
- 3 TC (Epivir, lamivudina, GR109714X)
- d4T (Zerit, estavudina)
- Abacavir (Ziagen, 1592U89)
- No nucleósidos (IRTNNs):
- Nevirapina (Viramune)
- Delavirdina (Rescriptor, U-90152T)
- Efavirenz (Sustiva)
- Saquinavir (Invirasa)
- Indinavir (Crixivan)
- Ritonavir (Norvir)
- Nelfinavir (Viracept)
- Amprenavir (Agenerase)
Los IRT alteran la función de la enzima llamada reverso transcriptasa, que es la que utiliza el VIH para cambiar su mensaje genético a una forma que pueda ser fácilmente insertada dentro del núcleo de la célula infectada. Siendo la encargada de hacer una copia en ADN del ARN viral, de manera que este pueda insertarse en el ADN celular mediante la acción de las integrasas, y así producir proteínas del VIH necesarias para la autorreproducción viral. Los IRTNs necesitan ser fosforilados para luego interactuar con un sustrato en el sitio de unión a la enzima y cuando es incorporado entonces termina la elongación de la cadena de ADN; mientras que los IRTNNs no necesitan ninguna conversión metabólica y bloquean directamente la reacción con la reverso transcriptasa, por una interacción específica con el sitio de unión "sin sustrato" de la reverso transcriptasa del VIH-1.2
Los IP son una clase de fármacos con una potente actividad antiviral. La proteasa del VIH es requerida para el clivaje del polipéptido gagpol en sus partes funcionales y la inhibición de este clivaje trae como consecuencia una disminución en la producción de viriones maduros. O sea, estos medicamentos se parecen a los trozos de la cadena de proteína que la proteasa corta normalmente. Al adherirse a la proteasa, los IP previenen que la proteasa corte las cadenas largas de proteínas y enzimas en trozos más cortos que el virus necesita para reproducirse en nuevas copias de sí mismo. Aunque las cadenas largas no hayan sido cortadas en los trocitos correctos, las nuevas copias del VIH aún se construyen y continúan empujando a través de la membrana de la célula infectada. Pero estas nuevas copias del VIH son "defectuosas" (ya que no han sido formadas completamente, por lo tanto no pueden continuar infectando otras células).3
Inhibidores de la reverso transcriptasa nucleósidos
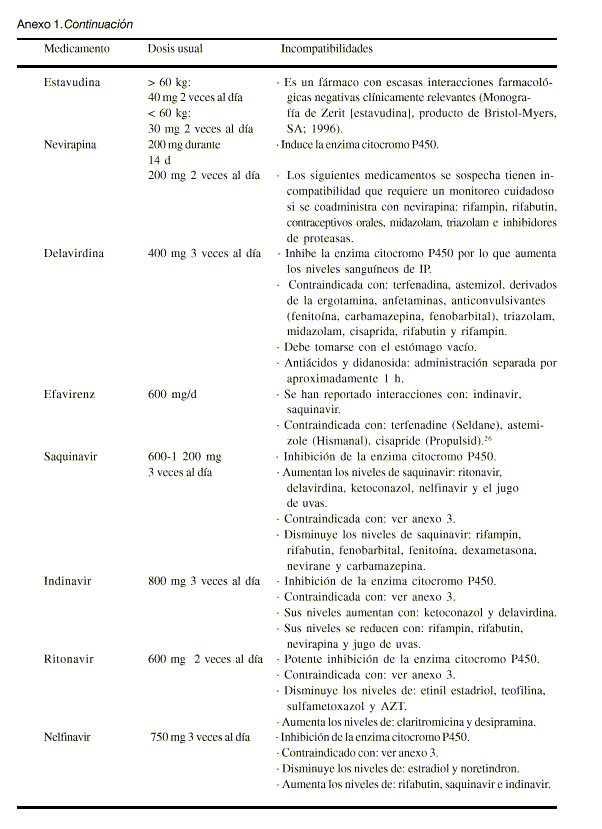
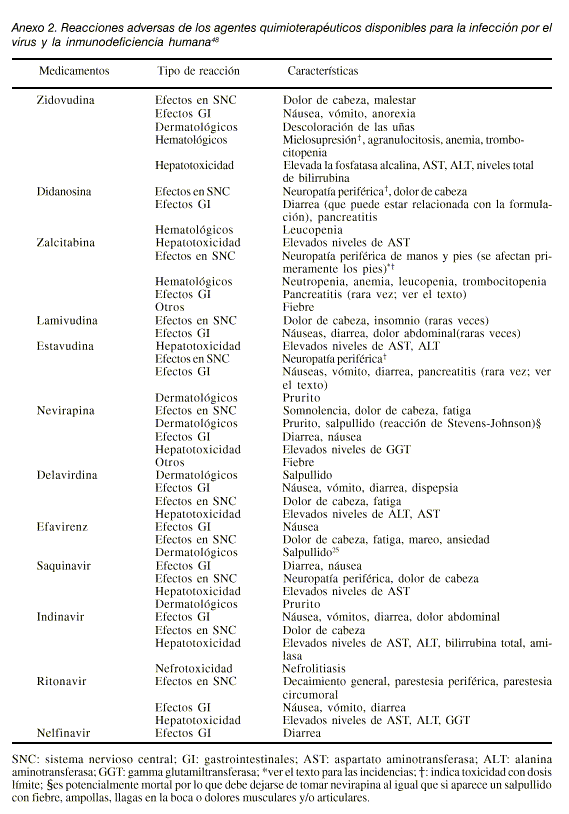
Las dosis, incompatibilidades y reacciones adversas de todo el grupo se presentan en los anexos 1 y 2.Zidovudina (aprobado en 1987)
El trifosfato de zidovudina (TZDV) es incorporado dentro del ADN viral por bloqueo competitivo de la reverso transcriptasa purificada del VIH, pero el fármaco sin fosforilar carece de actividad contra esta enzima.4,5 Zidovudina es también activa contra infecciones por virus Epstein-Barr (VEB) y por virus hepatitis B (VHB). El TZDV inhibe competitivamente la ADN polimerasa del VHB de una manera similar a la inhibición de la reverso transcriptasa vista en retrovirus.6 El mecanismo de acción de la zidovudina en la infección por VEB no está muy claro pero no se encuentra relacionado aparentemente con el efecto citotóxico del medicamento en cultivos celulares.7 El efecto protector de la zidovudina contra la citopatogenicidad inducida por el VIH puede verse bloqueada en relación con la dosis por la timidina, aunque la timidina no tiene efecto citotóxico inherente en las células infectadas por VIH.4,6,8,9 La zidovudina es metabolizada en el hígado por glucuronidación a 3' -ácido--3'-deoxi-5'-b -D-glucopiranosil timidina (GZVD),10 el cual es excretado en la orina por secreción tubular aunque en menor cantidad que el AZT (18 para 22 % GZVD contra 57 para 72 % de AZT) después de la administración por vía oral e intravenosa.11Didadosina (aprobado en 1991)
Didanosina es un nucleósido análogo sintético del nucleósido natural deoxya-denosina en el cual el grupo 3' -hidroxil (OH) es remplazado por hidrógeno. Intracelu-larmente, didanosina es convertido por las enzimas celulares al metabolito activo, dideoxiadenosina 5' -trifosfato (ddATP). El ddATP inhibe la actividad de la reverso transcriptasa del VIH-1 tanto por competición con el sustrato natural, deoxyadenosina 5' -trifosfato (dATP), como por su incorporación dentro del ADN viral. La falta del grupo 3' -OH en el nucleósido análogo incorporado previene la formación de fosfodiéster de unión de 5' a 3' esencial para la elongación de la cadena de ADN y, por tanto, se interrumpe el crecimiento del ADN viral. Para evitar una reducción en la biodisponibilidad de didanosina, este debe ser administrado al menos 30 min antes de las comidas (Prospecto de Videx) [didanosine], producto de Bristol-Myers Squibb Company, July, 1996).Zalcitabina (aprobado en 1992)
La zalcitabina es un análogo sintético del nucleósido natural 2' -desoxicitidina en el que el grupo 3'-hidroxilo ha sido sustituido por hidrógeno. Dento de las células, zalcitabina se transforma por mecanismos enzimoleculares en el metabolito activo: didesoxicitidin-5' trifosfato (ddCTP). El ddCTP actúa de sustrato alternativo en lugar del desoxicitidin-trifosfato (dCTP) de la reversotranscriptasa del VIH y, de este modo, por inhibición competitiva de la síntesis del ADN vírico, bloquea la replicación del VIH mediante la terminación prematura de la cadena. Pero tanto la velocidad de absorción como la concen-tración plasmática máxima disminuyen cuando se toma con alimentos y se duplica el tiempo hasta la concentración máxima (Prospecto de Hivid? 0.750 [zalcitabina], producto de F. Hoffmann- La Roche Ltd. Octubre, 1995). La neuropatía periférica (35 %) es la reacción adversa más comúnmente observada. Se caracteriza por una distribución en manos y pies, primeramente en los pies; está relacionada con la dosis; y usualmente aparece en los primeros 6 meses de terpia.12Lamivudina (aprobado en 1995)
Lamivudina es un nucleósido análogo de la citosina usado en el tratamiento de la infección causada por VIH-1 y/o por el virus de la hepatitis B (VHB).13 Tiene una potente actividad antiviral porque es resistente al clivaje desde el terminal 3' de los complejos ARN/ADN por la 3'-5' exonucleasa y no está sujeto a la desaminación. La actividad antiviral de lamivudina contra VIH-1 y VHB está dada por su anabolito activo 5' -trifosfato. El componente inicial es preferentemente anabolizado intracelularmente en mononucleocitos de sangre periférica en reposo. Inicialmente la droga es convertida a la forma de monofosfato por la deoxicitidin kinasa, luego pasa a la forma difosfato por la citidin monofosfato kinasa y deoxicitidin monofosfato kinasa, para finalmente pasar a la forma trifosfato activa por la pirimidin nucleósido difosfato kinasa.14 El difosfato de lamivudina es el anabolito predominante y se asume que sea el paso limitante en la fosforilación al trifosfato de lamivudina activo. Debe ser administrado en ausencia de alimentos.Estavudina (aprobado en 1994)
Es un análogo del nucleósido timidina. Es un profármaco que debe ser metabolizado dentro de las células por kinasas celulares a su forma trifosfato para ser activo. Al igual que el resto de los didesoxinucleósidos, penetra en el interior de las células infectadas donde se fosforila.15,16La estavudina inhibe la replicación del VIH por 2 mecanismos:
- Inhibe la actividad de la reverso transcriptasa del VIH, al competir con el sustrato natural, trifosfato de desoxitimidina.17
- Inhibe la síntesis del ADN viral al ser un terminal de cadena.
Se ha observado que el medicamento ha inducido pancreatitis la cual, aunque raras veces (1 %), ha provocado algunas muertes.
Abacavir
Abacavir (1592U89) es un análogo del nucleósido natural 2" desoxiguanosina que ha demostrado tener un perfil favorable y seguro en tratamientos clínicos iniciales en pacientes con VIH-1.18 Ha sido reportada resistencia cruzada in vitro con ddl, ddC y 3TC.19 Llega al sistema nervioso central (SNC), por lo que se está probando fundamentalmente en la demencia asociada con SIDA. No inhibe la cyt P450 y se ha probado tanto en niños como en adultos.En estudios realizados en niños ha sido bien tolerado. La reacción adversa más común fue rash y la dosis usada de 4-8 mg/kg de peso cada 12 h.20 En otros estudios realizados en adultos también fue bien tolerado a dosis entre 100 y 1 200 mg al día, pero la dosis de tratamiento más usada es de 300 mg cada 12 h. Las reacciones adversas más encontradas en este estudio fueron: astenia moderada, dolor abdominal, dolor de cabeza, diarrea y dispepsia.21
Inhibidores de la reverso transcriptasa no
Nucleósidos
La dosis, incompatibilidades y reacciones adversas de todo el grupo se presentan en los anexos 1, 2 y 3.Nevirapina (aprobado en 1996)
Nevirapina fue el primer IRTNNs aprobado por la Federal Drug Administration (FDA) para el tratamiento de adultos infectados con VIH. Es rápidamente absorbido después de su administración por vía oral. Debe tomarse siempre como parte de un régimen de combinación, nunca solo. Nevirapine provoca un rápido descenso en la carga viral, sin embargo, la aparición de resistencia ocurre a las pocas semanas de una monoterapia continua.22,23 El empleo previo de nevirapina puede afectar o eliminar la eficacia del uso subsecuente de efavirenz y delavirdina.
Delavirdina (aprobado en 1997)
El mesilato de delavirdina es un IRT del VIH-1 altamente específico que no se ha desarrollado grandemente para el tratamiento del SIDA.24,25 Delavirdina resulta más eficaz al ser combinado con análogos nucleósidos que no se hayan empleado previamente. Algunos investigadores recomiendan el empleo de la combinación de delavirdina/indinavir más 2 análogos nucleósidos para las personas cuyo régimen anterior con indinavir haya resulatdo insuficiente.Efavirenz (aprobado en 1998)
Efavirenz es un IRTNNs el cual muestra buena actividad inhibitoria contra el VIH-1.La reducción de la susceptibilidad al efavirenz ha sido reportada en variantes del VIH-1 que contienen mutaciones simples y múltiples de la enzima reverso-transcriptasa. Datos obtenidos sugieren que el perfil de resistencia del efavirenz se solapa con el de los IRTNNs nevirapina y delavirdina.26
Inhibidores de proteasas
La dosis, incompatibilidades y reacciones adversas de todo el grupo se presentan en los anexos 1, 2 y 3.Saquinavir (aprobado en 1995)
Fue el primer IP aprobado por la FDA para el tratamiento de adultos infectados por VIH. No se han publicado tratamientos en niños. Presenta un límite de biodisponibilidad luego de la dosis por vía oral del 4 % y es metabolizado por el sistema enzimático cyt P4503A.27-29 Debe tomarse dentro de las 2 h siguientes a la toma de alimentos.Indinavir (aprobado en 1996)
El indinavir se une al sitio activo de la proteasa e inhibe la actividad de la enzima. Esta inhibición previene el clivaje de las poliproteínas virales resultando en la formación de partículas virales inmaduras noinfecciosas (Crixivan, Indinavir sulfate. Resgitered trademark of Merck & Co., Inc., 1996). Debe administrarse en ausencia de alimentos.Ritonavir (aprobado en 1996)
Ritonavir es un IP con un perfil de resistencia al VIH-1 similar al del indinavir, pero diferente del saquinavir.30 Ritonavir inhibe la proteasa aspártica del VIH. Esta enzima es primordial en el procesamiento de productos de los genes gag y gag-pol en el funcionamiento de enzimas virales y del corte a proteínas del VIH. La inhibición resulta en la liberación de partículas virales no infecciosas inmaduras.31,32 Debe administrarse con comidas.Nelfinavir (aprobado en 1997)
Nelfinavir es un inhibidor de la proteasa del VIH-1. La inhibición de la proteasa viral previene el clivaje de la poliproteína gag-pol resultando en la producción de partículas virales inmaduras no infecciosas (Viracept, Nelfinavir mesylato. Registered trademark of Agouron Pharmaceuticals, Inc., 1998). Debe ser administrado con comidas.33Amprenavir
Amprenavir (141W94, VX-478, Agenerase) es un inhibidor de proteasa de segunda generación. El fármaco ha demostrado resultados alentadores durante los estudios clínicos humanos, y parece ser un inhibidor potente de la replicación del VIH. Los datos iniciales indican que amprenavir es capaz de penetrar la barrera cerebrovascular y el SNC, lo cual sugiere que el fármaco puede resultar beneficioso para las personas que padecen trastornos cognitivos relacionados con la demencia por VIH. En general, amprenavir parece ser bien tolerado. Al igual que los IP aprobados, amprenavir es metabolizado mediante la enzima cyt P450. No presenta resistencia cruzada in vitro con saquinavir e indinavir y sí en bajos niveles con ritonavir y nelfinavir. Se ha probado tanto en adulto como en niños. Se administra en dosis de 600 mg cada 12 h (4 cápsulas de 150 mg cada 12 h). Los efectos adversos más comunes son: náusea, vómito, diarrea, gases, fatiga, dolor de cabeza, salpullido y sensaciones de hormigueo alrededor de la boca (parestesia circunmoral).34 Se metaboliza mediante la enzima cyp 3A4 (cyt P450 3A4), por lo que interacciona con los fármacos expuestos en el anexo 3.35Antirretrovirales en investigación
Inhibidores de la reverso transcriptasa nucleósidos
Tenofovir (pmpa)
Es un nucleósido análogo con mucha potencia. Puede administrarse solo una vez al día. No se une al L-carnitina, eliminando la necesidad de suplementación de este. En estudios de fase I/II ha demostrado actividad clínica significativa.36Lodanosina (fdda)
Es un análogo nucleósido fluorado del ddl. Se encuentra en estudios fase I/II. Para su activación requiere ser fosforilado a FddATP, ya que su metabolito fosforilado tiene un tiempo de vida media (t?) largo, se piensa que es factible una sola dosis diaria. La ventaja en particular del FddA es que, a diferencia del ddl, es ácido estable, por lo que no tiene la necesidad de ser neutralizado, lo cual es una de las principales causas de los efectos adversos asociados con el ddl.36
FTC
Es una versión fluorada del 3TC, que parece ser más potente que este in vitro. Las características farmacocinéticas sugieren que puede ser conveniente la frecuencia de una vez al día. Resultados de estudios de fase I han demostrado una potente actividad antiviral in vivo y buena tolerancia.37Foziduvina tidoxil
Es un medicamento nucleósido análogo contra el VIH, experimental, similar a AZT que se encuentra en estudio de fase I.Lobucavir
Es un nucleósido análogo experimental. Efectivo contra diferentes clases de virus herpes, así como contra el VIH. También está siendo estudiado como un tratamiento contra el citomegalovirus (CMV).
Inhibidores de la reverso transcriptasa no nucleósidos
Atevirdina
Las bisheteroarilpiperazinas (BHAPs) son potentes IRTNNs del VIH-1 y el mesilato de atevirdina es el primero de esta clase y es estructuralmente diferente al resto de los IRTNNs disponible. Se ha demostrado que la atevirdina tiene una significativa actividad anti-VIH in vitro.38,39Atevirdina actúa en un sitio diferente de donde actúa IRTN como: AZT, ddl ó ddC. In vitro, los BHAPs muestran sinergia con AZT, ddl y ddC.40 Es bien tolerada a dosis entre 400- 1 200 mg.41
Trovirdina
Los componentes de las feniletiltiazoltiourea (FETT) representan una nueva clase de IRTNNs42 que inhiben preferentemente la reverso transcriptasa del VIH-1, pero no son inhibidores de otras ADN polimerasas. Estudios de cinética enzimática han mostrado que componentes de las FETT como el trovirdina actúa directamente sobre la reverso transcriptasa del VIH-1 y presentan una unión a esta, la cual difiere de las demás uniones de nucleósidos trifosfatos y ácidos nucleicos.43 Los componentes de las FETT comparten muchos rasgos comunes con otros IRTNNs,44 pero despliegan una mayor potencia anti-VIH.Lovirida (R 89439) es un IRTNNs que pertenece a la clase de los derivados de las a -AFA (alfa anilinofenilacetamida).2
MKC 422 es un IRTNNs con un nucleósido análogo en su centro; en estudios de fase I/II se ha asociado con importantes descenso en la carga viral.36
S-1153 se ha reportado que presenta in vitro más potencia que nevirapina y delavirdina, y al igual que el efavirenz se requiere de más de una mutación para que ocurran altos niveles de resistencia cruzada.
Carboxanilidas tienen un tiempo de vida largo pero requieren generalmente de administración por vía parenteral.
PNU 242721 es una tiopirimidina caracterizada por escasa unión a proteínas y se está estudiando la administración de 1 ó 2 veces al día.
Otros IRTNNs incluyen al HBY097 y al Canolide A, análogos que se encuentran en estudios de fase I.
Inhibidores de proteasas
Lopinavir (abt-378)
Es un nuevo inhibidor de proteasa, el cual cuando se combina con bajas dosis de ritonavir (11 mg 2 veces al día) provoca entre 50-100 veces aumento en la biodisponibilidad y prolongación del tiempo de vida media (t?), aproximadamente hasta 24 h. El aumento en los niveles de ABT-378 es específico para el ritonavir, pero no se ve con otros inhibidores de proteasas o con inhibidores de la cyt P450. Se encuentra en estudios fase II.36KNI-272 (Kynostaatin). Inhibidor de proteasa contra el VIH, experimental. Se encuentra en estudios de fase I en niños y adultos.
Otros inhibidores de proteasa en estudios en fase temprana incluyen: tipranivir (PN 140690E), la ciclourea DMP-450, PD-178390 y el péptido BMS-232632.
Se encuentran en desarrollo varios nuevos agentes antirretrovirales que actúan en otros sitios del ciclo de vida viral:
- Los inhibidores de integrasa están siendo estudiados, pero su desarrollo ha sido lento por su escasa potencia y toxicidad no deseada.
- Los antagonistas de los receptores de las kimoquinas tienen el potencial para bloquear la entrada del VIH a la célula. Algunos candidatos de esta línea han sido estudiados, pero se han encontrado ciertos problemas: como el hecho de que algunos aislamientos de VIH son comunes para más de un tipo de receptor de kimoquinas; que la actividad de algunos agentes es dependiente del tipo de célula y que hay cierta incertidumbre acerca de la toxicidad a largo plazo por bloqueo de los receptores.
- El primer inhibidor de la fusión, T-20 (DP-178), se encuentra en ensayos clínicos. T-20 se une a la gp41 en la envoltura viral e inhibe la fusión entre el VIH y la célula. Esto al parecer constituye una actividad potente pero requiere de administración por infusión o inyección subcutánea.36
- Los inhibidores de los dedos de zinc (CI-1012) son agentes que inhiben áreas del VIH denominadas de igual forma "dedos de zinc" los cuales tienen una serie de funciones en el ciclo de vida viral, pero son especialmente impor-tantes en el ensamblaje de nuevos virus, según van saliendo de una célula infectada. Cuando los "dedos de zinc" son bloqueados, el VIH hace copias de sí mismo que no funcionan y no pueden infectar nuevas células.36
- Los liposomas de conexión a CD4+ son usados como trampa para bloquear la replicación del VIH por unión a la gp 120 del VIH.
CONCLUSIONES
Los avances recientes en el tratamiento de combinación ofrecen nuevas esperanzas a las personas VIH+. El tratamiento con regímenes de 3 fármacos ha producido mejoras clínicas, mayor tiempo de supervivencia y ha aumentado la calidad de vida de muchas personas. Estas potentes combinaciones anti-VIH se conocen como el tratamiento antirretroviral sumamente activo o TARSA. Ejemplo de los regímenes TARSA incluyen: 1 inhibidor de proteasa más 2 análogos nucleósidos o un análogo no nucleósido más 2 nucleósidos análogos, etc. La FDA, de los Estados Unidos de Norteamérica, es decir, el organismo que regula los medicamentos, decidió en lo que a drogas para combatir el SIDA respecta, terminar con la burocracia y dar rápida aprobación a aquellos medicamentos provenientes de laboratorios prestigiosos.En este trabajo se da información de los principales antivirales que ya están en ensayos clínicos o en uso y brindan al médico la posibilidad de dar una más eficiente atención a estos pacientes. Se hace además un bosquejo de las reacciones adversas y de las incompatibilidades de cada uno de los medicamentos nombrados, dando una elección de acuerdo con las características individuales de estos pacientes.
Anexo 1. Dosis usuales e incompatibilidades de los agentes quimioterapéuticos disponibles para la infección por el virus de la inmunodeficiencia humana 45

Anexo 2. Reacciones adversas de los agentes quimioterapéuticos disponibles para la infección por el virus y la inmunodeficiencia humana48
Anexo 3. Medicamentos que pueden interactuar potencialmente con los inhibidores de proteasas vía cyt P450, específicamente la isoenzima cyp3A4 del metabolismo

SUMMARY
During the last years many new antiviral agents have been incorporated to the chemotherapeutics. The pharmaceuticals established years ago as well as the new ones developed to treat HIV infected individuals are included in this review. The AZT was the first approved in March, 1987, followed by ddl (1991), ddc (1992), d4t (1994), and 3TC (1995). Later, the first protease inhibitor, saquinovir, was approved in December, 1995, followed by ritonavir (1996), indinavir (1996), and nelfinavir (1997); in addition to other inhibitors of the reverse transcriptase as neviparine (1996), delavirdine (1997), and efavirenz (1998), among others. At present new alternative therapies for this affection are being searched and developed. Some of the characteristics of these dugs, such as: action mechanisms (on which enzime each of them act and how they do it, viral cycle), dosage, incompatibilites and adverse reactions are dealt with in this paper.Subject headings: ANTIVIRAL AGENTS/drug therapy; ACQUIRED IMMUNODEFICIENCY SYNDROME/pharmacolopgy; DRUG THERAPY, COMBINATION.
REFERENCIAS BIBLIOGRÁFICAS
- Havlir DV, Lange JM. New antiretrovirals and new combinations. AIDS 1998;12(Suppl A): S165-74.
- De Clercq E. What can be expected from Non-nucleoside reverse transcriptase inhibitors (NNRTIs) in the treatment of human immunodeficiency virus type 1 (HIV-1) infections?. Med Virol 1996;6:97-117.
- Markowitz M. Los inhibidores de la proteasa. Chicago: Abbott Laboratory, 1997:
- Nordenfelt E, Lofgren B, Chattopadhyaya J, Oberg E. Inhibition of hepatitis B virus ADN polimerase by 3'-azido-3'-deoxythymidine triphosphate but not its threo analog. J Med Virol 1987;22:231-6.
- Vrang L, Bazin H, Remaud G. Inhibition of the reverse transcriptase from HIV by 3'-azido-3'-deoxythymidine triphosphate and its threo analogue. Antiviral Res 1987;7:139.
- Nakashima H, Matsui T, Harada S. Inhibition of replication and citopathic effect of human T cell lymphotropic virus type III/lymphadenopathy associated virus by 3'-azido-3'-deoxythymidine in vitro. Antimicrob Agents Chemother 1986;30:933-7.
- Lin J-C, Zhang ZX, Smith MC, Biron K, Pagano JS. Anti-human Immunodeficiency virus agent 3'-azido-3'--deoxythymidine inhibits replication of Epstein-Barr virus. Antimicrob Agents Chemother 1988;32:265-7.
- Sommadossi JP, Carlisle R, Schinazi RF, Zhou Z. Uridine reverses the toxicity of 3'-azido-3'-deoxythymidine in normal human granulocyte-macrophage progenitor cells in vitro without impairment of antiretroviral activity. Antimicrob Agents Chemother 1988;32:997-1001.
- Eriksson B, Vrang L, Bazin H. Different patterns of inhibition of avian myeloblastosis virus reverse trasncriptase activity by 3'-azido-3'-deoxythymidine 5'-triphosphate and its threo isomer. Antimicrob Agents Chemother 1987;31:600-4.
- Miranda P de, Burnette TC, Good SS. Disposition and pharmacokinetics of the antiviral drug 3'-azido-3'-deoxythymidine (Retrovir) in monkeys and rats. Proceeding of 27th Interscience Conference on Antiviral Agents and Chemotherapy. New York, October 4-7, 1987: (Abstract No. 378):162.
- Sim SM, Back DJ, Breckenridge AM. The effect of various drugs on the glucuronidation of zidovudine 8azidothymidine; (AZT) by human liver microsomes. Br J Clin Pharmacol 1991;32:17-21.
- Blum AS, Dal Pan GJ, Feingberg J. Low-dose zalcitabine-related toxic neurophaty: frecuency natural history, and risk factors. Neurology 1996;46:999-1003.
- Johnson MA, Moore KHP, Yuen GJ. Clinical Pharmacokinetics of lamivudine. Clin Pharmacokinet 1999;36(1):41-66.
- Cammack N, Pouse P, Marr CLP, et al. Cellular metabolism of (-) -enantiomeric 2'-deoxy-3'-thiacytidine. Biochem Pharmacol 1992;43(10):2059-64.
- August EM, Birks EM, Prusoff W. 3'-Deoxythymidin-2'-enepermeation of human lymphocyte H9 cells by nongfacilitated difusion. Mol Pharmacol 1991;39:246-9.
- Domin BA, Mahony WB, Zimmerman TP. 2', 3'-Dideoxythymidine permeation of the human erythrocyte membrane by nonfacilitated diffusion. Biochem Biophys Res Commun 1988;154(3):825-31.
- Huang P, Farguhar D, Plunkett W. Selective action of 2', 3'- didehydro-2', 3'-dideoxythymidine triphosphate on human immunodeficiency virus reverse transcriptase and human DNA polimerases. J Biol Chem 1992;267(4): 2817-22.
- Hughes W, McDowell JA, Shenep J. Safety and single-dose pharmacokinetics of abacavir (1592U89) in human immunodeficiency virus type 1-infected children. Antimicrob Agents Chemother 1999;43(3):609-5.
- Foster RH, Faulds D. Abacavir. Drugs 1998;55(5):729-36.
- Hughes W, McDowell JA, Shenep J, Flynn P, Kline MW, Yogev R, et al. Safety and single-dose pharmacokinetics of abacavir (1592U89) in human immunodeficiency virus type 1-infected children. Antimicrob Agents Chemother 1999;43(3):609-15.
- Kumar PN, Sweet DE, McDowell JA, Symonds W, Lou Y, Hetherington S, et al. Safety and pharmacokinetics of abacavir (1592U89) following oral administration of escalating single doses in human immunodeficiency virus type 1-infected adults. Antimicrob Agents Chemother 1999;43(3):603-8.
- Richman DD, Havlir D, Corbeil J. Nevirapine resistance mutations of human immunodeficiency virus type 1 selected during therapy. J Virol 1994;68:1660-6.
- Havlir D, Cheeseman SH, McLaughlin M. High-dose nevirapine: safety, pharmacokinetics, and antiviral effect in patients with human immunodeficiency virus infection. J Infec Dis 1995;171:537-45.
- Chang M. Identification of the metabolites of the HIV-1 reverse transcriptase inhibitor delavirdine in monkeys. Drug Metab Dispos 1997;25(7):814-27.
- Chang M. Metabolism of the HIV-1 reverse transcriptase inhibitor delavirdine in mice. Drug Metab Dispos 1997;25(7):828-39.
- Adkins JC, Noble S. Efavirenz. Drugs 1998;56(6):1055-64.
- Kitchen VS, Skinner C, Ariyoshi K. Safety and activity of saquinavir in VIH infection. Lancet 1995;345:952-5.
- Noble S, Faulds D. Saquinavir. A review of its pharmacology and clinical potential in the management of VIH infection. Drugs 1996;52:93-112.
- Schapiro JM, Winters MA, Stewart F. The effect of high-dose saquinavir on viral load and CD4+ T-cell counts in VIH-infected patients. Ann Intern Med 1996;124:1039-50.
- Lea AP, Faulds D. Ritonavir. Drugs 1996;52(4):541-6.
- Moyle G, Gazzard B. Current knowledge and future prospects for the use of HIV protease inhibitors. Drugs 1996;51(5):701-12.
- Kempf DJ, Marsh KC, Denissen JF. AT-538 is a potent inhibitor of human immunodeficiency virus protease and has high oral bioavailability in humans. Proc Natl Acad Sci USA 1995;28(92):2484-8.
- Guía de tratamientos experimentales. La Red de Información del SIDA, 1997. [Disponible en]: http://www.aidsnyc.org/lared/index. html.
- Adkins JC, Faulds D. Amprenavir. Drugs 1998;55(6):837-42.
- Decker CJ, Laitinen LM, Bridson GW, Raybuck SA, Tung RD, Chaturvedi PR. Metabolism of amprenavir in liver microsomes: role of CYP3A4 inhibition for drug interactions. J Pharm Sci 1998;87(7):803-7.
- Deeks S, Barditch-Crovo P, Lietman PS. The safety and efficacy of PMPA prodrug monotherapy: preliminary results of a phase I/II dose-escalation study. 5th Conference on Retroviruses and Opportunistic Infections, Chicago, 1998 (abstract LB8). [Disponible en]:http://hiv.medscape.com/Medscape/HIV/Treatment/Update/1998/tu01/eng/pnt-eng.tu01.html.
- Pottage J, Thompson M, Kahn J, Delehanty J, McCreedy B. Potent antiretroviral efficacy of low dose FTC, initial results from a phase I/II clinical trial. 5th Conference on Retroviruses and Opportunistic infections, Chicago, 1988 (abstract LB9). [Disponible en]:http://hiv.medscape.com/Medscape/HIV/Treatment/Update/1998/tu01/eng/pnt-eng.tu01.html.
- Romero DL, Busso M, Tan CK, Reusser F, Palmer JR, Poppe SM, et al. Nonnucleoside reverse transcriptase inhibitors that potently and specifically block human immunodeficiency virus type 1 replication. Proc Natl Acad Sci USA 1991;88:8806-10.
- Althaus IW, Chou JJ, Gonzales AJ, Deibel MR, Chou KC, Kezdy FJ, et al. Steady-state kinetic studies with the non-nucleoside HIV-1 reverse transcriptase inhibitor U-87201E. J Biol Chem 1993;268:6119-24.
- Campbell TB, Young RK, Eron JJ, D?Aquila RT, Tarpley WG, Kuritzkes DR. Evaluation of the bisheteroarylpiperazine U-87201E in combination with zidovudine of didanosine against human immunodeficiency virus type 1 infection. J Infect Dis 1993;169:318-26.
- Been-Tiktak AMM, Vrehen HM, Schneider MME, Feltz M van der, Branger T, Ward P, et al. Safety, tolerance, and pharmacokinetics of Atevirdine Mesylate (U-87201E) in asymptomatic human immunodeficiency virus-infected patients. Antimicrob Agents Chemother 1995;39(3):602-7.
- Ahgren C, Bäckbro K, Bell FW, Cantrell AS, Clemens M, Colacino J, et al. A new class of potent nonnucleoside inhibitor of human immunodeficiency virus type 1 reverse transcriptase. Antimicrob Agents Chemother 1995;39(6):1329-35.
- Zhang H, Vrang L, Bäckbro K, Lind P, Sahiberg C, Unge T, et al. Inhibition of wild type and mutant reverse transcriptase by the phenyl ethyl thiazolyl thiourea derivates trovirdine and MSC-127. Antiviral Res 1995;28:331-42.
- Young SD. Non-nucleoside inhibitors of HIV-1 reverse transcriptase. Perspect Drug Discovery Design 1993;1:181-92.
- Bartlett JG. Pocket Book of infectious Disease Therapy. 9 ed. Baltimore: Williams and Wilkins, 1998:203-6.
- Kewn S, Veal GJ, Hoogard PG. Lamivudine (3TC) phosphorylation and drugs interactions in vitro. Biochem Pharmacol 1997;54:589-95.
- Moore KH, Yuen GJ, Raasch RH. Pharmacokinetics of lamivudine administered alone and with trimetoprim-sulphametoxazole. Clin Pharmacol Ther 1996;59(5):550-8.
- Beach JW. Chemotherapeutic agents for human immunodeficiency virus infection: mechanism of action, pharmacokinetics, metabolism, and adverse reactions. Clin Ther 1998;20(1):2-25.
Lic. Alicia Tarinas Reyes. Instituto de Medicina Tropical "Pedro Kourí". Autopista Novia del Mediodía, Km 6 1/2, Marianao 13, P.O. Box 601, Ciudad de La Habana, Cuba.
1Licenciada en Ciencias Farmacéuticas. Aspirante a Investigadora.
2Licenciado en Química. Investigador Titular.
3Especialista de II Grado en Farmacología. Investigador Auxiliar.

