










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Farmacia
versión impresa ISSN 0034-7515versión On-line ISSN 1561-2988
Rev Cubana Farm v.35 n.3 Ciudad de la Habana sep.-dic. 2001
Diseño y validación de un método espectrofotométrico para el estudio de estabilidad de Benzocaína en un nuevo ungüento rectal
Yania Suárez Pérez,1 Oscar García Pulpeiro2 y Fathel Mohamed Ahmed Al-Sufi3
Resumen
Se desarrolló un método por espectrofotometría UV, que incluyó la extracción previa del producto de degradación de la benzocaína presente en un nuevo ungüento rectal, destinado al tratamiento del brote hemorroidal agudo. El método analítico se validó para el estudio de estabilidad química de este principio activo. Según los criterios estadísticos vigentes, el método resultó lineal, exacto, preciso, sensible y específico para el objetivo con el cual fue diseñado.
DeCS: TECNOLOGIA FARMACEUTICA; ESTABILIDAD DE MEDICA-MENTOS/métodos; composición DE MEDICAMENTOS; ESPECTRO- FOTOMETRIA ULTRAVIOLETA; BENZOCAINA/farmacología; QUIMICA FARMACEUTICA; POMADAS/uso terapéutico.
La Industria Farmacéutica se encuentra involucrada en garantizar la máxima calidad posible de los productos desde la etapa de diseño, cumpliendo con las llamadas Buenas Prácticas de Fabricación y Laboratorio. Para ello es necesario tener en cuenta una serie de aspectos que incluyen la calificación de equipos y del personal, la documentación y la validación de las técnicas o métodos de análisis.
La validación de un método analítico incluye la evaluación de una serie de parámetros que dependerán del estudio en que será utilizada la técnica en cuestión. Ellos son: linealidad, precisión, exactitud, especificidad y sensibilidad.1
La benzocaína (p-amino benzoato de etilo) es el éster etílico del ácido p-aminobenzoico (PABA). El PABA a su vez es el principal producto de degradación de la benzocaína por hidrólisis. Su perfil de pH revela catálisis básica, aunque también puede esperarse que presente catálisis ácida. De ahí se deduce que para incrementar la estabilidad de la benzocaína, se debe minimizar su contacto con agua, bases y ácidos.2
En la bibliografía se reportan muy pocas técnicas específicas que permiten la determinación de la benzocaína en presencia del PABA. Entre ellas se destacan la espectrofotometría diferencial basada en la capacidad del PABA de sufrir efecto batocrómico en zona alcalina. Otro consiste en acomplejar benzocaína con cafeína después de aplicar hidrólisis alcalina, mientras que la solución acuosa de PABA se determina en la zona UV.2
Entre estos métodos se incluyen lógicamente técnicas cromatográficas. Se reportan 2 técnicas por cromatografía líquida de alta resolución con adecuada sensibilidad y selectividad que garantizan la separación y cuantificación simultánea de benzocaína y PABA.2 Otras técnicas reportadas más recientemente incluyen la cuantificación de benzocaína en presencia de otros principios activos.3-5
La cromatografía gaseosa se utilizó con éxito en el estudio de estabilidad de un ungüento de uso tópico, empleando detector de ionización de llama.6
El ungüento rectal que contiene este fármaco es un producto en etapa de investigación, que requiere de los estudios de estabilidad para poder ser registrado. Por esta razón, nos encaminamos a desarrollar un método útil para este fin, que pudiera satisfacer los requisitos actuales de validación estipulados para los métodos de la categoría II.7
Métodos
Reactivos
- Cloroformo p.a.
- Etanol 96° p.n.
- Sulfato de sodio anhidro.
Equipos
- Balanza analítica digital ER - 60 A.
- Balanza analítica digital Sartorios 1720.
- Espectrofotómetro UV-VIS. Pye -Unicam SP6-450.
- Espectrofotómetro UV-VIS. Pharmacia LKB.
Desarrollo de un método específico para cuantificar benzocaína en presencia de su producto de degradación. Teniendo en cuenta las solubilidades relativas de la benzocaína y de su producto de degradación, se diseñó un método por espectro-fotometría UV que incluyó la extracción previa del PABA en fase acuosa, por separación en sistemas de 2 fases. La benzocaína se determinó en fase clorofórmica y el PABA se eliminó en fase acuosa.
Determinación de la capacidad de eliminación de interferencias del PABA en la fase clorofórmica con la aplicación de lavados acuosos sucesivos. La solubilidad relativa del PABA en agua con respecto a la benzocaína es muy superior.
Se preparó una solución de PABA en agua 3 mg/mL de la cual se tomaron 50 mL y se mezclaron en un embudo separador con igual volumen de cloroformo. Se agitó y se separó la fase clorofórmica. De cada fase se tomaron 100 µL y se enrasó a 10 mL con etanol 96 °.
Se determinó la absorbancia a 295 nm contra un ensayo de corrección.
La misma solución clorofórmica se sometió a lavados sucesivos con 4 fracciones de 50 mL de agua. Después de cada extracción se colectaron 100 µL y se enrasaron en matraces de 10 mL con etanol 96 °. Las soluciones finales se leyeron a 295 nm. Con estos resultados se construyó un gráfico de absorbancia vs número de lavado para cada fase y se observó si el procedimiento resultó eficaz o no.
Descripción del método
- Se pesaron con exactitud 300 mg de ungüento en un beacker de 50 mL.
- Se añadieron 20 mL de cloroformo, se agitó para disolver la benzocaína y se trasvasó a un embudo separador de 125 mL.
- Se lavó el beacker con 10 mL de cloroformo y se trasvasó al embudo.
- Se extrajo el PABA de la fase clorofórmica mediante el lavado con 4 fracciones sucesivas de 50 mL de agua.
- Se desechó la fase acuosa y se colectaron los extractos clorofórmicos en un beacker de 100 mL.
- Se añadieron cantidades apropiadas de sulfato de sodio anhidro para secar la fase clorofórmica con ayuda de agitación.
- Se filtró por papel rápido hacia volumétricos de 50 mL.
- Se completó el volumen con cloroformo.
- Se extrajo una alícuota de 2 mL y se trasvasó a un volumétrico de 50 mL.
- Se completó el volumen con cloroformo.
- Se determinó la absorbancia a 280 nm contra ensayo de corrección.
Forma de cuantificación utilizada para estimar el contenido de benzocaína en el ungüento. Se utilizó la comparación con el estándar externo. Se preparó una solución de benzocaína en cloroformo de concentra-ción 4,8 µg/mL, la cual se evaluó en iguales condiciones que las muestras y se determinaron los valores de absorbancia contra ensayo de corrección a 280 nm. El cálculo de la concentración experimental en tanto por ciento se realizó mediante la expresión siguiente:
Cm =Am ´ Cp/Ap
donde:
Cm = concentración que se desea estimar (%).
Am = absorbancia de la muestra a 280 nm.
Cp= concentración del estándar externo (100 %).
Ap = absorbancia del estándar externo a 280 nm.
Validación del método espectrofo-tométrico con extracción previa para estudios de estabilidad. El método se validó teniendo en cuenta los parámetros exigidos para la categoría II que agrupa a los métodos analíticos para determinación cuantitativa del fármaco sin interferencias de sus productos de degradación en las formas terminadas.
Los parámetros evaluados fueron:
- Especificidad.
- Especificidad en estabilidad.
- Linealidad del sistema.
- Linealidad del método.
- Exactitud.
- Precisión: repetibilidad y reproduci-bilidad.
- Sensibilidad: Límite de detección y de cuatificación.
Especificidad. La especificidad con respecto a los excipientes se evaluó mediante el proceso por triplicado de placebos del producto por la técnica propuesta. Se analizó la presencia de respuesta positiva o no de los componentes de la matriz en el rango de interés analítico para la benzocaína. Además se registró el espectro UV contra blanco de cloroformo entre 245 y 330 nm.
Especificidad en estabilidad. Se procesaron por triplicado placebos cargados con 15 mg de PABA (aproxima-damente 3 veces la cantidad de benzocaína presente en la porción de ensayo) y se determinó la absorbancia de la fase clorofórmica a 280 nm, la que se comparó con el estándar externo equivalente al 100 % de benzocaína en cloroformo.
Además se analizaron por triplicado placebos cargados simultáneamente con 6 mL de solución madre de benzocaína en cloroformo 1 mg/mL, es decir, la cantidad equivalente al 100 % y con 15 mg de PABA. Finalizado el procesamiento, se determinó el tanto por ciento de recobro de benzocaína (R) y se comparó nuevamente con el estándar externo preparado al 100 % de la dosis. Se verificó la posibilidad de interferencia del PABA en los porcentajes de benzocaína obtenidos.
Linealidad del sistema. Cumplimiento de la ley de Lambert-Beer. Para evaluar el cumplimiento de la ley de Lambert-Beer se construyó una curva de calibración de absorbancia vs concentración, utilizando diferentes diluciones de benzocaína en cloroformo correspondientes a 2,4; 3,6; 4,8; 6,0 y 7,2 µg/mL. Estas diluciones correspon-dieron a las concentraciones de 50, 75, 100, 125 y 150 % del fármaco en relación con el contenido nominal en el ungüento. Cada punto se evaluó por duplicado. Los datos se procesaron estadísticamente con la aplicación de la regresión lineal mediante el paquete STATISTICA. Se determinó r (coeficiente de correlación lineal), r2 (coeficiente de determinación), intercepto (a) y pendiente (b) para el 95 % de confianza. Se aplicaron los criterios de aceptación siguientes:
- Ecuación de la recta: y = bx + a
- r ³ 0,99
- r2 ³ 0,98
- Prueba de proporcionalidad del método analítico o hipótesis nula de la ordenada en el origen a = 0. Se empleó la prueba estadística t de Student para n_2 grados de libertad siendo n el número total de pares de valores; donde t experimental < t teórica.
- Prueba de la hipótesis nula de la pendiente b = 0. Se determinó a partir de una prueba ANOVA de la regresión, o mejor, la probalidad (p) asociada con el valor de la pendiente, es decir, si la p<< 0,05 el valor de "b" difiere significativamente de cero.
Linealidad del método, exactitud y repetibilidad. Para la evaluación de estos 3 parámetros se utilizaron los mismos datos. Se llevó a cabo con placebos cargados con el 50, 75, 100, 125 y 150 % de benzocaína. La preparación de las muestras consistió en pesar individualmente 300 mg de placebo, a los cuales se les adicionaron alícuotas de 3; 4,5; 6; 7,5 y 9 mL respectivamente de una solución madre de benzocaína en cloroformo 1 mg/mL. Cada punto se procesó por triplicado.
Se aplicaron los criterios de aceptación siguientes:
- Linealidad del método: Se procedió de la misma forma descrita para la evaluación de la linealidad del sistema; de manera que se construyó la curva de calibración de concentración experi-mental (%) vs concentración teórica (%) y se aplicó el mismo procesamiento estadístico y los mismos criterios de aceptación.
- Exactitud: Se construyó la curva de recuperación que incluyó los valores obtenidos para 3 niveles de concentración: bajo (75 %), medio (100 %) y alto (125 %). Estos datos se procesaron por regresión lineal de igual modo que para la linealidad, por lo que deben cumplirse los mismos criterios de aceptación. Además se calculó para cada punto el tanto por ciento de recobro (R), el recobrado medio (R) y el coeficiente de variabilidad total (CV).
(R) = Y/X
donde:
Y = absorbancia de la muestra.
X= absorbancia del estándar externo.
R = 96-104 %
CV £ 3,0 %
- Repetibilidad: Se analizaron las réplicas de los 3 niveles de concentración utilizados para evaluar la exactitud. Se calculó para cada punto el CV. Estas determinaciones se realizaron por el mismo analista en las mismas condiciones de trabajo. CV £ 2,0 %.
Reproducibilidad. Se cargaron placebos del producto con alícuotas de 6 mL de una solución madre de benzocaína en cloroformo 1 mg/mL. Participaron 2 analistas en 2 días diferentes, quienes analizaron 3 réplicas cada uno para un total de 12 ensayos. Los resultados se evaluaron estadísticamente mediante un análisis de varianza.
El criterio de aceptación fue: CV£ 3,0 %.
Sensibilidad: Límite de detección y de cuatificación. Para la evaluación de este parámetro se estimaron el límite de cuantifi-cación (Lc) y el límite de detección (Ld) a partir de la curva de regresión aplicando los pasos siguientes:
- Se determinó la pendiente de la curva de calibración de la linealidad del sistema: concentración (µg/mL vs absorbancia) y se obtuvo el valor de "b".
- Se construyó otra curva de calibración analizando 3 puntos con 3 réplicas cada uno de concentraciones menores de analito (0,8, 1,2 y 1,6 µg/mL) y se determinó la ecuación de esta nueva recta de calibración aplicando regresión lineal con el paquete STATISTICA. Se extrapoló la respuesta a concentración cero y se obtuvo un estimado de la respuesta blanco "Ybl".
- Se determinó la desviación estándar (S) correspondiente a cada concentración evaluada en el paso anterior. Se construyó otra curva de calibración de concentración (µg/mL) vs desviación estándar (Sx). Al igual que en el paso anterior, se extrapoló S a concentración cero y se obtuvo el estimado "Sbl" correspondiente a la S del blanco.
Se calculó Lc y Ld mediante las ecuaciones siguientes:
Ld = (Ybl + 3Sbl)/b x (1 / Ö n)
Lc = (Ybl + 10Sbl) / b x (1 /Ö n)
donde:
Ld: límite de detección.
Lc: límite de cuantificación.
b: pendiente de la recta de regresión lineal.
Ybl: estimado del blanco.
Ysl: estimado de la desviación estándar del blanco.
n: número de medidas individuales.
Se comprobaron de forma experimental los resultados obtenidos teóricamente para ambos límites, preparando por triplicado las soluciones correspondientes de benzocaína en cloroformo en cada caso.
Resultados
Se determinó que la longitud de onda de máxima absorción de la benzocaína 4,8 µg/mL en cloroformo fue a 280 nm, por lo que se fijó este valor para llevar a cabo la determinación.
Además se verificó que los valores de absorbancia permanecieron estables durante 8 h.
En la figura 1 se muestra la capacidad del método para eliminar las posibles interferencias debidas al PABA de la fase clorofórmica, en la cual se lleva a cabo la cuantificación del fármaco.
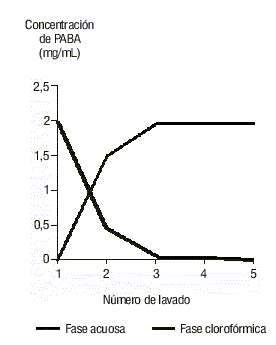
En dicha figura se evidenció una rápida transferencia del PABA a la fase acuosa a partir del segundo lavado. El producto de degradación estuvo totalmente ausente después de la cuarta extracción, por lo que de este modo se justifica la selección de este número de lavado en la descripción del método. Si se tiene en cuenta que la cantidad de PABA que se añadió equivale aproximadamente al 250 % con respecto a la degradación total de la benzocaína en concentraciones ideales, podemos brindar adecuada seguridad en relación con la eficacia de este paso.
El primer parámetro analizado en la validación del método fue la especificidad. La figura 2 muestra que el método es específico frente a las sustancias auxiliares y el otro fármaco incluido en la formulación, ya que no se obtuvo respuesta en el rango de interés analítico para la benzocaína.
Los mismos resultados con respecto a los valores de absorbancia se obtuvieron al cargar placebos del producto con cantidades apreciables de PABA, por lo que tampoco existieron interferencias desde el punto de vista cualitativo, debidas al principal producto de hidrólisis en la fase clorofórmica. Desde el punto de vista cuantitativo, los resultados también fueron satisfactorios pues la cuantificación de la benzocaína en los placebos cargados simultáneamente con el principio activo y el PABA, dieron valores de recobrados adecuados entre 99 y 101 %, para un valor medio de 99,39 % ± 0,87. De este forma quedó demostrada no solo la especificidad para control de calidad, sino también la especificidad del método para estudios de estabilidad.
Se verificó la linealidad del sistema, es decir, el cumplimiento de la ley de Lambert--Beer en el rango entre 2,4 y 7,2 µg/mL. Para este mismo rango, pero trabajando en lugar de patrones con placebos cargados, se comprobó la linealidad del método. Para ambos parámetros se alcanzaron elevados coeficientes de correlación lineal y determinación. Para el intercepto el valor de t de Student experimental fue menor que el teórico para el 95 % de confianza y para los grados de libertad utilizados en cada caso, por lo que el intercepto no difiere significativamente de cero. Por otra parte, se mostraron pendientes próximas a la unidad, con t de Student muy altas y valores de probabilidad (p) inferiores al 0,05 establecido como límite, por lo que podemos concluir que el valor observado es prácticamente igual al predicho. El cumplimiento de todos los criterios estadísticos de la regresión avaló la linealidad del sistema y del método.
Con los resultados de los 3 puntos centrales de la linealidad del método se evaluó la exactitud y la repetibilidad. Para la exactitud el procesamiento estadístico de la curva de recuperación cumplió con las exigencias establecidas al igual que en la linealidad. Este resultado nos condujo a considerar que los errores sistemáticos del método no fueron significativos, por lo que puede considerarse exacto. Aunque para el recobrado medio ( R) ningún valor excedió los límites de 97-103 % propuestos por la mayoría de los autores, consideramos la ampliación de estos en ± 1 % (96-104 %) tal como se recomienda para las preparaciones semisólidas. A su vez el coeficiente de variación total fue muy inferior al 3,0 % estipulado como límite.
La repetibilidad a los 3 niveles dio valores de coeficiente de variación bajos, los cuales cumplen satisfactoriamente con el 2,0 % propuesto como límite (tabla 1).
Los resultados de la reproducibilidad se muestran en la tabla 2. Como se observa el coeficiente de variación total es menor que el 3,0 %. Además se comprobó por el programa ANOVA-2 que no existen diferencias significativas desde el punto de vista estadístico entre los resultados obtenidos para días (F = 0,402) y analistas (F = 0,345), pues en ambos casos la F calculada resultó inferior a la tabulada. De este modo, pudimos concluir que el método fue repetible y reproducible, es decir, fue suficientemente preciso, por lo que no repercutieron apreciablemente los errores aleatorios en la determinación.

Fig. 2. Resultados del ensayo de especificidad. Espectro UV del placebo procesado por el método propuesto.
Por último se determinó la sensibilidad. La tabla 3 resume los resultados obtenidos para el límite de detección y cuantificación, así como los resultados experimentales alcanzados en la comprobación de los límites.
Como hemos demostrado, el método cumplió con los parámetros mínimos necesarios para ser aplicado en el seguimiento de la estabilidad química de la benzocaína en este producto. Además de ser lineal, exacto, preciso y específico frente a las sustancias auxiliares, fue suficientemente sensible y permitió diferenciar la respuesta de la benzocaína y de su producto de degradación.
Discusión
La benzocaína es prácticamente insoluble en agua y muy soluble en cloroformo, al igual que los excipientes utilizados en la formulación, ya que se utilizó una base oleaginosa. El PABA es soluble en agua, por lo que consideramos oportuno realizar su extracción en medio acuoso a partir de la fase apolar, con el objetivo de lograr adecuada especificidad del método frente al producto de degradación. Además se evaluó la especificidad frente a la matriz, su objetivo fue demostrar la ausencia de interferencias de los restantes componentes de la formulación. La cuantificación de benzocaína por espectrofotometría UV directa, es decir, sin extracción previa del PABA, carece de valor para el seguimiento de la estabilidad química del fármaco y solo puede ser recomendada para el control de calidad en el producto recién elaborado. El método utilizado garantiza seguridad y selectividad frente a todos los componentes de la matriz.
Tabla 1. Resultados de la evaluación de los parámetros linealidad del sistema, linealidad del método, exactitud y repetibilidad
| Conc. Real (%) | Linealidad del sistema Absorbancia a 280 nm | Linealidad del método Conc. Experimental (%) Y ± DE | Exactitud Recobrado (%) Y ± DE | Repetibilidad Coeficiente de variación (%) |
| 50 |
0,27 ± 0,00 | 49,53 ± 0,462 | - | - |
| 75 |
0,42 ± 0,63 | 74,93 ± 0,414 | 100,23 ± 1,086 | 1,080 |
|
100 | 0,56 ± 0,00 | 99,99 ± 1,815 | 99,99 ± 1,815 | 1,815 |
|
125 | 0,71 ± 0,00 | 122,51 ± 0,525 | 97,70 ± 0,854 | 0,875 |
|
150 | 0,86 ± 0,00 | 149,90 ± 1,153 | - | - |
|
Resultados | Y = 1,22 x-0,023 | Y = 0,9892 x+ 0,249 | Y = 9,948 x+ 0,45 | - |
|
|
R2 = 0,9999 | R2 = 0,9990 | R2 = 0,9979 | |
|
R = 0,9999 | R = 0,9980 | R = 0,9959 | ||
|
Texp (0,13) <tteo | texp (0,19)<tteo | texp(1,96)<tteo | ||
| p= 0,000 |
p = 0,000 | p= 0,000 | ||
|
R = 99,31 % | ||||
|
CV = 1,67 % | ||||
|
Criterios | Y =bx+a | Y = bx+a | Y =bx+a | CV £2,0 % |
|
R2 ³ 0,99 | R2 ³ 0,99 | R2 ³ 0,99 | ||
|
r ³ 0,98 | r ³ 0,98 | r ³ 0,98 | ||
| a @ 0: |
a @ 0: | a @ 0: | ||
|
texp<tteo | texp<tteo | texp<tteo | ||
|
b@ 1 | b@ 1 | b@ 1 | ||
|
p<<0,05 | p<<0,05 | p<<0,05 | ||
|
R = 96-104 % | ||||
|
CV £ 3,0 % |
Tabla 2. Resultados de la reproducibilidad del método
| Días |
Analista 1 | Analista 2 |
|
1 | 97,58 | 96,77 |
|
96,77 | 96,54 | |
|
96,77 | 97,00 | |
| 2 |
97,20 | 96,77 |
|
97,00 | 97,40 | |
|
96,77 | 96,80 | |
|
Resultado: CV = 0,358 % | Criterio: CV £ 3,0 % | |
La demostración de la linealidad del sistema permite establecer en el rango estudiado una proporcionalidad directa entre la concentración de analito y la respuesta medida, que es la absorbancia en este caso. Si bien este resultado es muy importante, no debemos limitarnos a estimar la linealidad solo para el sistema, ya que las características de la matriz en estudio y el trabajo experimental realizado con el desarrollo del método, puede provocar algunas modificaciones en los resultados con respecto al procedimiento mucho más simplificado que se lleva a cabo con los patrones.
Con respecto a la exactitud no se confrontaron dificultades con el cumplimiento de los criterios. Sin embargo, la disminución de los valores de R con el aumento de la concentración pudiera parecer contradictoria. Estos resultados pueden ser perfectamente explicados si partimos de la base de que en los 30 mL de cloroformo añadidos al inicio se disuelve la totalidad de la benzocaína, y a partir de esa concentración se realizan las 4 extracciones con agua. Las pequeñas pérdidas de volumen que ocurren en esta etapa, repercuten marcadamente en la concentración final. Aunque las pérdidas sean mínimas, si la muestra estaba más concentrada, se observa menor valor de R.
Tabla 3. Resultados del ensayo para la determinación de la sensibilidad
|
Concetración µg/mL | Absorbancia a 280 nm (X ± DE) |
|
0,8 | 0,0786 ± 0,061 |
|
1,2 | 0,1200 ± 0,056 |
|
1,6 | 0,1700 ± 0,010 |
|
Pendiente de la recta | 0,12229 |
|
Intercepto Yhl | 0,00000 |
|
Intercepto Sbl | 0,01200 |
|
Límite de detección | 0,203 µg |
|
Límite de cuantificación | 0,677 µg |
| Comprobación experimental de los límites absorbancia a 280 nm | |
| Límite de detección | 0,020 |
| 0,019 | |
| 0,020 | |
| Límite de cuantificación | 0,070 |
| 0,069 | |
| 0,070 | |
La precisión y la sensibilidad de la técnica fueron satisfactorias, por lo que el método es válido para el objetivo con el cual fue diseñado. Mediante la técnica propuesta se puede cuantificar pequeñas cantidades de fármaco, en el orden de los microgramos con elevada especificidad.
Summary
A method including the previous extraction of the degradation product of benzocaine pesent in a new rectal ointment to treat acute haemorrhoid was developed by ultraviolet spectrophotometry. The analytical method was validated for the study of the chemical stability of this active principle. According to the standing statistical criteria, the method proved to be lineal, exact, accurate, sensitive and specific for the objective it was designed.
Subject headings: TECHNOLOGY, PHARMACEUTICAL; DRUG STABILITY//methods; DRUG COMPOUNDING; SPECTROPHOTOMETRY, ULTRAVIOLET; BENZOCAINE/pharmacology; CHEMISTRY, PHARMACEUTICAL; OINTMENTS/therapeutic use.
Referencias bibliográficas
- M. Gil-Alegre. Protocolos de validación de métodos de análisis, materias primas y productos terminados. Madrid: Universidad Complutense, 1995:26-32.
- Connors, KA Amidon GL, Kennon Ll. Chemical Stability of Pharmaceuticals. A handbook for pharmacists. New York: John Wiley, 1979:175-8.
- Reed. GH, Evaluation of electrochemical detection in reversed phase HPLC. J High Resolut Chromat Common 1988:11(9):675-7.
- Gigante B, Barros AM, Teixeira A, Marcelo-Curto MJ. Separation and simultaneous high _performamce liquid chromatographic determination of benzocaine and benzol benzoatina pharmaceutical preparation. J Chrom 1991;549(2):217-20.
- Caraballo L, Fernández A, Holgado MA, Vela M, Rabasco AM. Rapid HPLC method for the quantification of tyothricin, menthol and benzocaine in pharmaceutical formulation. J Pharm Sci 1994;83(8):1174-9.
- Biemer TA, Asral N, Alabanese IA. Simultaneous stability indicanting capillary gas chromatographic assay for benzocaine and two principal benzil esters of Balzam Peru formulated in a topical oitment J. Chrom 1992:623(2):395-8.
- United State Pharmacopoeial Convention. USP 23. United State Pharmacopoeia. 23 ed. Easton: Mack Printing,1995:1982-4.
Recibido: 23 de febrero del 2001. Aprobado: 30 de mayo del 2001.
M. Sc. Yania Suárez Pérez. Palatino No. 53 entre Salvador y Meireles, municipio Cerro, Ciudad de La Habana, Cuba.
1 Máster en Tecnología y Control de Medicamentos. Instituto de Farmacia y Alimentos (IFAL). Universidad de La Habana.
2 Máster en Tecnología y Control de Medicamentos. Empresa Laboratorio "Roberto Escudero Díaz". Industria Médico Farmacéutica. (IMEFA).
3 Licenciado en Ciencias Farmacéuticas.

