










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Farmacia
versión impresa ISSN 0034-7515versión On-line ISSN 1561-2988
Rev Cubana Farm v.39 n.3 Ciudad de la Habana sep.-dic. 2005
Instituto de Farmacia y alimentos. Universidad de La Habana
Diseño y validación de un nuevo método para estimar reductores hidrosolubles asociados con la quitina
Yania Suárez Pérez,1 Oscar García Pulpeiro2 y Marisel Cueto León3
Resumen
Se desarrolló por primera vez un método espectrofotométrico para estimar reductores hidrosolubles asociados con la quitina materia prima. Para el ajuste del método se utilizó glucosamina como sustancia de referencia. Las determinaciones se realizaron a 330 nm que fue la longitud de onda de máxima absorción. Se seleccionó como tiempo óptimo de calentamiento 5 min y se verificó la selectividad de la respuesta analítica. Se demostró el cumplimiento de la ley de Lambert-Beer en el intervalo de 100-300 mg/mL. Se obtuvo un extracto acuoso a partir de quitina materia prima procesado para eliminar posibles interferencias, el cual se empleó como control en la validación del método. El método fue lineal, exacto y preciso en el intervalo estudiado. El límite de detección fue de 25,21 mg/mL y el de cuantificación de 65,45 mg/mL.
Palabras clave: Quitina, estabilidad, productos de hidrólisis, glucosamina, espectrofotometría.
La quitina es un fármaco de probada estabilidad química y térmica1,2 utilizado ampliamente en la terapéutica por sus efectos farmacológicos y como excipiente.3
El estudio cinético realizado a la quitina cubana se realizó en hidrolizados ácidos cuantificando los azúcares reductores por la técnica de Schoorl.4 Se obtuvo un t10 % de 134 x 104 años, lo cual indica la alta resistencia del polímero a la degradación.4
Los posibles productos de degradación final de la quitina por vía hidrolítica son: Qquitosana, N-acetilglucosamina, glucosamina y ácido acético. Estos productos, solo se obtienen en condiciones muy drásticas de concentración de álcali y de ácidos minerales fuertes, catalizada por altas temperaturas o por acción enzimática.5,6 Los productos de degradación por ruptura de los enlaces glicosídicos b 1 ® 4, oligómeros de diferente peso molecular, son los responsables de los efectos biológicos, ya que la quitina es un profármaco. No obstante, desde el punto de vista de estabilidad química, se impone demostrar que la quitina no sufre modificaciones durante el almacenamiento.
Teniendo en cuenta estos aspectos, se propone un método analítico que se destina a evaluar la presencia de oligómeros hidrosolubles de diferente tamaño molecular, que acompañan a la quitina desde su proceso de obtención. Estos productos solubles en agua, pueden tener el grupo amino libre o acetilado, por lo que no es recomendable realizar la cuantificación en base solo a estos grupos. Sin embargo, independientemente de la acetilación, se generan por la ruptura de los enlaces glicosídicos b(1 ® 4), grupos formilos libres, responsables del carácter reductor de estos compuestos de diferente tamaño molecular.
El desarrollo de un método capaz de determinar el contenido de estos compuestos reductores presentes en la fracción hidrosoluble obtenida a partir de la quitina, permite establecer un criterio adicional sobre la estabilidad química del polímero. Con este objetivo se evaluó un nuevo método espectrofotométrico para cuantificar el contenido de reductores presentes en fase acuosa procedentes de la quitina, expresado como glucosamina, la cual se utilizó como sustancia de referencia.
Métodos
Materias primas. Quitina polvo. Fabricante Empresa Mario Muñoz. Cuba. Lote 9801 CM: F 590006050.
Reactivos. Hidróxido de sodio BDH, ácido clorhídrico Merck, etanol 96 ° pn, glucosamina Merck.
Desarrollo del método. Se evaluó un método basado en determinar espectrofotométricamente el compuesto formado, al someter la muestra a un medio fuertemente alcalino aportado por el hidróxido de sodio (NaOH). En la figura 1 se describe el procedimiento aplicado; para el análisis se empleó la glucosamina como sustancia de referencia.
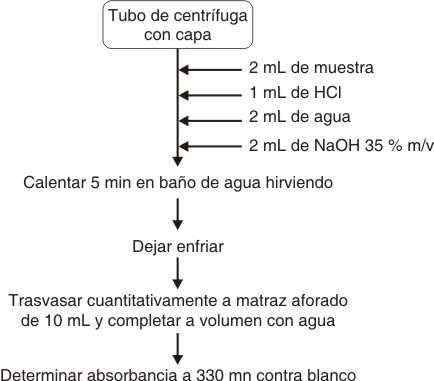
Fig. 1. Método espectrofotométrico desarrollado para glucosamina sustancia de referencia.
Ensayos realizados para glucosamina sustancia de referencia
Determinación de la l máxima. Se preparó una solución de glucosamina en agua 1 mg/mL. Se aplicó el procedimiento descrito en la figura 1 y se registró el espectro UV en el intervalo de 280-380 nm vs blanco. Las determinaciones se realizaron en un espectrofotómetro UV-VIS Pye Unicam SP6-450 (Inglaterra). Se determinó la l de máxima absorción.
Determinación de la influencia del tiempo de calentamiento en la absorbancia. Se evaluaron muestras de glucosamina preparadas de igual forma a la empleada en el ensayo anterior y se aplicó el procedimiento de la figura 1. Se varió el tiempo de calentamiento (1, 3, 5,10, 15, 20 y 30 min) y se determinó la absorbancia a la l de máxima absorción.
Estabilidad de la absorbancia a la l de máxima absorción . Se evaluó la misma muestra recién preparada y al transcurrir 5, 10, 15, 20, 30, 45 y 60 min.
Evaluación de la selectividad del método. Se realizaron los espectros UV de 270 a 400 nm de la solución de NaOH 35 % m/v, de glucosamina patrón 0,2 mg/mL en presencia o no de NaOH de igual concentración.
Determinación del cumplimiento de la ley de Lambert-Beer. Se realizó una curva de calibración de absorbancia a la l de máxima absorción vs concentración (mg/mL). Se emplearon 5 niveles de concentración de glucosamina. Se partió de una solución de glucosamina 1 mg/mL en agua de la cual se tomaron alícuotas de 1; 1,5; 2; 2,5 y 3 mL, correspondientes al intervalo 100-300 µg/mL. Cada punto se analizó por triplicado y los resultados se procesaron por regresión lineal con el empleo del programa Statistica versión 6.1.
Obtención del extracto acuoso a partir de quitina materia prima
Teniendo en cuenta que la aplicación del método se realizó para el análisis de la fracción hidrosoluble obtenida a partir de la quitina, fue necesario establecer las condiciones para obtener el extracto acuoso.
Se pesó con exactitud 1 g de quitina materia prima y se colocó en un tubo de centrífuga con tapa, al cual se le añadió 6 mL de agua desionizada. Se tapó el tubo y se agitó vigorosamente hasta lograr total humectación de la quitina. Se agitó el tubo en zaranda durante 30 min y se filtró cuantitativamente sobre papel, desechando el residuo.
Una vez fijadas las condiciones, se procedió a introducir un paso en el procedimiento. Se incorporó la hidrólisis durante 1 h con 1 mL de HCl 0,1 N, colocando la muestra (previo al desarrollo de coloración con NaOH 35 % m/v), en un baño de agua hirviendo. Se dejó enfriar y se continuó de la forma que se muestra en la figura 1. Este paso tiene como objetivo favorecer la hidrólisis de los enlaces glicosídicos ß 1®4 de los oligómeros de diferente tamaño presentes en la fracción hidrosoluble.
Evaluación de la influencia de la hidrólisis . Se determinó el espectro UV a 0,5 mL de extracto acuoso obtenido a partir de la quitina en presencia o no de HCl, con calor o sin calor durante 1 h, para evaluar el posible efecto de la hidrólisis. Se registró este entre 270-400 nm vs ensayo de corrección. Se realizaron 3 réplicas en cada caso.
Evaluación de la influencia del calentamiento en presencia de NaOH . Se repitió el ensayo anterior para muestras de 0,5 mL de extracto acuoso, en presencia de NaOH 35 % m/v, las cuales se sometieron o no al calentamiento durante 5 min. Los espectros se compararon con el obtenido para una muestra de extracto acuoso solo, a igual concentración.
Eliminación de interferencias del extracto acuoso obtenido a partir de quitina materia prima
Se obtuvo un extracto acuoso libre de interferencias en el intervalo de interés analítico para emplearlo como referencia en la posterior validación del método.
Se procedió a lavar sucesivamente 1 g de quitina con 6 mL de agua en un tubo de centrífuga con tapa. Se utilizaron iguales proporciones de quitina y agua que en el extracto. Se agitó vigorosamente y se centrifugó 10 min a 3 500 r/min. Se determinó la absorbancia de la muestra que contenía una alícuota de 0,5 mL del sobrenadante, procesada por el método en estudio. Se repitió el procedimiento hasta seleccionar el número de lavados que permitiera la eliminación de interferencias en la solución acuosa resultante.
Validación para estudios de estabilidad de la quitina materia prima
Se evaluaron los parámetros selectividad, linealidad del método, exactitud, precisión, límite de detección (Ld) y de cuantificación (Lc). Se emplearon controles (extracto acuoso libre de interferencias) cargados con glucosamina 1 mg/mL sustancia de referencia en el mismo intervalo analizado para la linealidad del sistema (100-300 µg/mL).
La linealidad del método se analizó con controles cargados con glucosamina en 5 valores de concentración en el mismo intervalo en que se evaluó el cumplimiento de la ley de Lambert-Beer. Los análisis se realizaron por triplicado. Los resultados se procesaron por regresión lineal con el empleo del programa Statistica versión 6.1.
La exactitud se evaluó mediante la curva de recuperación y el recobrado medio obtenido al cargar por triplicado placebos en 3 valores de concentración. En estos mismos niveles de concentración se evaluó la repetibilidad. La precisión intermedia se analizó a la concentración equivalente al 100 % con la participación de 2 analistas en 2 días diferentes.
La selectividad del método se comprobó analizando por triplicado controles del producto en presencia o no de glucosamina, sustancia de referencia equivalente al 100 % (200 µg/mL) por el método en estudio. Se registraron los espectros UV y se cuantificó el contenido de glucosamina por comparación contra patrón 100 %. Se determinó media y desviación estándar (DE).
Para la determinación de Ld y Lc se aplicó el procedimiento de estimación teórica descrito en la bibliografía a partir de la curva de calibración de la linealidad del método.7
Comprobación experimental del límite de cuantificación. Se procesaron por triplicado controles cargados con 65,5 µg/mL de glucosamina sustancia de referencia. Se cuantificó por el método en estudio y se calculó media y DE.
Resultados
En la figura 2 se resumen los resultados de las evaluaciones preliminares realizadas para glucosamina sustancia de referencia. Se seleccionó 330 nm por corresponder con la l de máxima absorción, y se demostró la estabilidad de esta en el tiempo. Se seleccionó 5 min de calentamiento, como tiempo óptimo en el estudio de su influencia en la respuesta analítica, teniendo en cuenta que se obtuvo la mayor intensidad en la absorbancia. Se verificó la selectividad de la reacción entre la glucosamina y el NaOH, ya que el analito no dio respuesta analítica y el NaOH no causó interferencias en el intervalo de interés analizado.
El cumplimiento de la ley de Lambert-Beer dio resultados satisfactorios. Según puede apreciarse, el procesamiento por regresión lineal demuestra el cumplimiento de los criterios de aceptación establecidos, lo cual garantiza la proporcionalidad directa entre concentración de analito y respuesta obtenida en el intervalo de 100-300 µg/mL.

Fig. 2. Resultados de los estudios preliminares realizados en el desarrollo del método espectrofotométrico para glucosamina sustancia de referencia: espectro UV de glucosamina con NaOH y selectividad de la reacción frente a NaOH (A), estabilidad de la absorbancia e influencia del tiempo de calentamiento (B) y cumplimiento de la ley de Lambert-Beer (C).
Una vez demostrada la posibilidad de aplicación del método para glucosamina y establecidas las condiciones para la reacción con el NaOH, se procedió a obtener un extracto acuoso a partir de la quitina materia prima.
Se empleó un procedimiento sencillo de modo que pudiera reproducirse fácilmente. Se obtuvo por filtración un extracto concentrado, ya que solo se añadieron 6 mL de agua por cada gramo de quitina. El extracto acuoso obtenido a partir de quitina materia prima, absorbió luz UV considerablemente en la zona de interés, en ausencia de NaOH (fig.3A). Es importante señalar que este extracto acuoso presentó coloración debido a impurezas generadas en el proceso de obtención de la quitina.
Con el objetivo de verificar si la composición del extracto acuoso se modifica, debido al proceso de hidrólisis, se realizó el tratamiento con HCl 1 N durante 1 h, calentando en agua hirviendo. En la figura 3 A se observa el espectro del extracto hidrolizado y sin hidrolizar. La presencia de HCl no produjo modificaciones en el espectro, con respecto a la del extracto acuoso al 100 % con calentamiento o sin este. No obstante, se mantuvo esta etapa por las posibilidades de variaciones en la composición de extractos obtenidos de diferentes lotes de quitina materia prima, que pueden ser analizados en el futuro. El incremento del contenido de reductores en la fracción hidrosoluble producto de la hidrólisis, conduciría a mejoras en la sensibilidad del método. Estos estudios deben ser realizados con posterioridad.
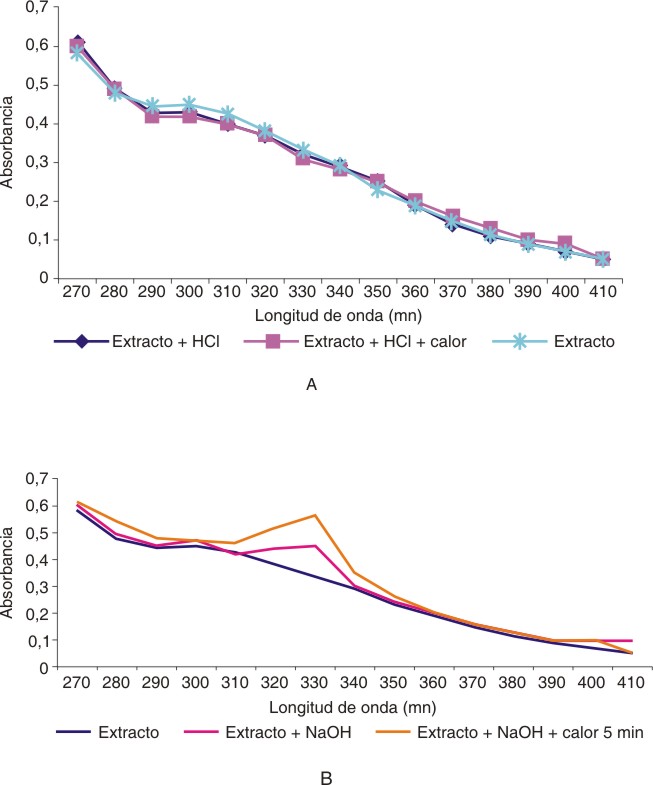
Fig.3. Evaluaciones espectrofotométricas realizadas con el extracto acuoso obtenido a partir de la quitina materia prima: influencia de la hidrólisis con HCl (A) y efecto de la reacción con NaOH en presencia de calor o no (B).
Al analizar el espectro de la muestra en presencia de NaOH, se observa un máximo a 330 nm (fig. 3 B). Los valores de absorbancia se incrementan apreciablemente, cuando se repite este ensayo con 5 min de calentamiento en baño de agua hirviendo. De esta forma se demostró que los extractos hidrosolubles obtenidos a partir de la quitina, contienen una pequeña concentración de compuestos reductores que responden selectivamente a la reacción en estudio. El calentamiento para aumentar la intensidad del color es ventajoso, ya que mejora apreciablemente la respuesta analítica. Si para iguales valores de concentración la absorbancia se incrementa, el método será más sensible en presencia de calentamiento. Este resultado es muy conveniente para estudios de estabilidad.
Para realizar la validación del método, se impuso trabajar con controles del producto. Si partimos del extracto acuoso obtenido a partir de la quitina, sin eliminar previamente las interferencias observadas, se afectarían los resultados. Por esta razón en la validación del método para materia prima, se empleó extracto acuoso obtenido a partir de quitina, previamente tratado hasta quedar libre de interferencias. Se comprobó que 5 lavados fueron suficientes para eliminar completamente las interferencias.
Se verificó la selectividad del método ya que los controles libres de interferencias no absorbieron en el intervalo en estudio. La recuperación media de glucosamina fue de 98,76 ± 1,24 %.
El cumplimiento de todos los criterios de aceptación establecidos, permitió demostrar que el método fue lineal, exacto y preciso en el intervalo de concentraciones de 100-300 µg/mL. Además se estimaron los Ld y de Lc (tabla).
Tabla. Resultados de la validación del método espectrofotométrico para determinación de reductores hidrosolubles en el extracto acuoso obtenido a partir de quitina materia prima
| Parámetro | Resultados | Criterios |
| Linealidad del método | Y =0,9415X+0,00977 r =0,9960 r 2 =0,9915 t exp < t tab (a =0,05, n=13) t Þ 1,9758 < 2,16 b = 0,9415 t = 40,6025 p = 0,0000 CV f =4,29 % | Y=bx+a r ³0,99 r2 ³0,98 Intercepto no significativo t exp < t tab (2,16) b @ 1 t alta p £ 0,005 CV f £ 5 % |
| Exactitud | Y=0,9802X+ 2,0633 r =0,9977 r 2 =0,9948 t exp < t tab (a =0,05; n=7) t Þ 0,7678< 2,36 b = 0,98023 t = 39,40 p = 0,0000 CV f = 2,304 % | Curva de recuperación Y=bx+a r =0,99 r2 =0,98 Intercepto no significativo t exp < t tab (2,36) b @ 1 t alta p = 0,005 CV f £ 5 % |
| R total = 100,61 % CV total = 1,96 % | R total = 97-103% CV total £ 3,0 % | |
| S2 max = 3,88 G exp = 0,3147 G tab = 0,8709 | Prueba de Cochran G exp < G tab µ= 0.05 K=3 n=3 | |
| t exp (0,3917)< t tab | Prueba de la t Student t exp < t tab ( 12,71 ) | |
| Repetibilidad | CV 50 % = 0,74 % CV 100 % =1,30 % CV 150 % =1,42 % | CV £1,5 % |
| Precisión intermedia | CV=1,00 % | CV total £ 3,0 % |
| F exp (1,272) < F tab t exp (0,672)< t tab | Fischer(F) y Student(t) entre analistas F tab = 5,05; t tab = 2,23 | |
| F exp (3,956) < F tab t exp (0,081)< t tab | Fischer (F) y Student (t) entre días F tab = 5,05; t tab = 2,23 | |
| Sensibilidad | Ld= 25,21µg/mL Lc= 65,45 µg/mL |
Se recomienda la aplicación del método a la determinación de reductores hidrosolubles asociados con la quitina materia prima, responsables de la caramelización por calor, fenómeno que afecta la calidad de esta.
Discusión
La aplicación del método espectrofotométrico se basa en la determinación de la absorbancia a la longitud de onda (l) de máxima absorción, del compuesto coloreado formado a partir de glucosamina sustancia de referencia, en medio fuertemente alcalino, aportado en este caso por el hidróxido de sodio. El desarrollo de este método es novedoso tanto desde el punto de vista de aplicación a glucosamina, como al seguimiento de la estabilidad química de oligómeros hidrosolubles asociados con la quitina. Se conoce del oscurecimiento de los azúcares en medio alcalino, sin embargo, se desconoce el mecanismo de la reacción, por lo que se debe estudiar posteriormente.
Aunque los oligómeros hidrosolubles no son productos tóxicos y su formación in situ es beneficiosa desde el punto de vista farmacológico; frente al calor son los responsables de la caramelización que conlleva al oscurecimiento de la materia prima. Por esta razón, consideramos de importancia su estimación como una prueba de control de calidad a la materia prima y como criterio adicional de la estabilidad del polímero. Esta prueba puede ser establecida como prueba límite de sustancias relacionadas en la materia prima.
Summary
Design and validation of a new method for estimation of water-soluble reducers associated with chitin
For the first time, a spectrophotometric method to estimate water-soluble reducers associated with raw material chitin was developed. For the purpose of adjusting the method, glucosamine as a reference substance was used. The estimations were made at 330 nm that was the wavelength of maximum absorption. The selected optimal heating time was 5 min; the selectivity of analytical response was checked. The Lambert-Beer law was demonstrated in the 100-300 mg/mL interval. An aqueous extract from raw material chitin was obtained and then processed to eliminate possible interference and be used as a control for the method validation. The method was linear, exact and accurate in the studied interval. The detection limit was 25,21 mg/mL and the quantification limit was 65,45 mg/mL.
Key words: Chitin, stability, hydrolysis products, glucosamine, spectrophotometry.
Referencias bibliográficas
1. García I, Oviedo D, Henriques RD. Influencia de las diferentes etapas de obtención y purificación en el comportamiento térmico de la quitina. Bol Inf Cient Tecn Inst Quin Biol Experiment. Academia de Ciencias de Cuba. 1982;2 (1):28-39.
2. García J, Henriques RD. Caracterización termoanalítica de la quitina. Bol Inf Cient-Tecn Inst Quim Biol Experiment. Academia de Ciencias de Cuba. 1981;165:1-11.
3. Kumar R. A review of Chitin and Chitosan applications. React Funct Polymer. 2000;46:1-27.
4. Nieto OM. Quitina. Su estudio y utilización como fármaco acelerador de la cicatrización. Tesis presentada en opción al grado científico de Doctor. en Ciencias Farmacéuticas. Universidad de La Habana. Instituto de Farmacia y Alimentos, 1993. p. 67-89.
5. Ekblad A, Nasholm T. Determination of chitin in fungi and mycorrhizal roots by an improved HPLC analysis of glucosamine. Plant Soil. 1996;178(1):29-35.
6. Duarte ML, Ferreira MC, Marvao MR, Rocha J. Determination of the degree of acetylating of chitin materials by
13 C CP/MAS NMR spectroscopy. Int J Biol Macromol. 2001;28:359-63.
7. ICH Harmonised Tripartite Guideline. Validation of analytical procedures: Methodology. Q2B. 1996. p. 7-8.
Recibido: 29 de junio de 2005. Aprobado: 2 de agosto de 2005.
Dr. Yania Suárez Pérez. Instituto de Farmacia y Alimentos. Universidad de La Habana. Ave 23 No. 21425 entre 214 y 222, La Coronela, La Lisa, Ciudad de La Habana. Correo electrónico: yania_as@yahoo.es
1Doctora en Ciencias Farmacéuticas. Profesora Asistente.
2Maestro en Tecnología y Control de Medicamentos. Laboratorio Roberto Escudero.
3Licenciada en Ciencias Farmacéuticas. Laboratorios MEDSOL.

