










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Farmacia
versión On-line ISSN 1561-2988
Rev Cubana Farm v.41 n.1 Ciudad de la Habana ene.-abr. 2007
Centro de Investigación y Desarrollo de Medicamentos
Método analítico por cromatografía líquida de alta resolución para el estudio de estabilidad del jarabe de loratadina 0,1 %
Leonid Torres Amaro,1 Caridad M. García Peña2 y Zenia Pardo Ruiz2
Resumen
Se validó un método de cromatografía líquida de alta resolución para el estudio de estabilidad del jarabe de loratadina 0,1 %. La curva de calibración en el rango de 13,6 a 3,36 µg/mL fue lineal, con un coeficiente de correlación igual a 0,99975; la prueba estadística para el intercepto y la pendiente, no significativa. El recobrado obtenido resultó de 100,2 % en el rango de concentración estudiado, y las pruebas de Cochran y Student (t), no significativas. El coeficiente de variación en el estudio de repetibilidad fue igual a 0,41 % para 10 réplicas ensayadas, mientras que en la reproducibilidad las pruebas de Fischer y Student, no significativas. El método resultó específico, lineal, preciso y exacto.
Palabras clave: Loratadina, cromatografía líquida de alta resolución, validación.
Loratadina es un derivado piperidínico de la azatadina, de acción antihistamínica con actividad central sedativa o antimuscarina. Es indicado para aliviar los síntomas de las reacciones de hipersensibilidad, incluyendo la rinitis alérgica y la urticaria crónica.1,2
Existen reportes de métodos analíticos por espectrofotometría ultravioleta para formulaciones de loratadina tabletas debido a que es un producto que absorbe intensamente en la región del ultravioleta.3 Para el jarabe de loratadina aparecen reportes de métodos analíticos por cromatografía líquida de alta resolución.4
La cromatografía líquida de alta resolución (CLAR) ha dado la posibilidad al analista de usar esta herramienta para solucionar los inconvenientes de los métodos espectrofotométricos en los estudios de estabilidad, pues además de presentar una alta sensibilidad y exactitud, es en esencia un método separativo; lo que permite medir con gran selectividad el compuesto deseado, siempre y cuando se encuentre un sistema cromatográfico que asegure una adecuada separación.4
La validación es el proceso establecido para la obtención de pruebas documentadas de que el método es lo suficientemente fiable para producir el resultado.5-8
En el presente trabajo se describe y valida un método analítico por CLAR a fin de cuantificar el principio activo para los estudios de estabilidad del jarabe de loratadina 0,1 %.
Métodos
La sustancia de referencia química de loratadina fue suministrada por el grupo de sustancias de referencia del Centro de Investigaci ón y Desarrollo de Medicamentos (CIDEM). El producto terminado en forma de jarabe al 0,1 %, fue elaborado en la Grupo de Formas terminadas, perteneciente al CIDEM. Todos los reactivos utilizados fueron de grado HPLC.
En el ensayo se empleó un cromatógrafo (KNAUER) con detector UV/VIS (KNAUER) ajustado a 254 nm, un dosificador (loop) de 20 µL e integrador (SHIMADZU CR6A), con una sensibilidad de 0,08 AUFS. La separación se realizó isocráticamente sobre una columna Licrospher RP 18, 250 mm x 4 mm, 10 µm. La fase móvil utilizada consistió en una mezcla filtrada y desgasificada de metanol: dihidrogenofosfato de potasio 0,013 mol/L, ajustada a pH 7,6-8,0 con hidróxido de potasio 0,1 mol/L en proporción 8/2 (v/v), con una velocidad de flujo de 2,0 µL/min. Todos los reactivos utilizados fueron de calidad analítica (MERCK, Alemania).
Con el objetivo de evaluar la especificidad del método cromatográfico, se sometió el producto terminado de la formulación seleccionada a oxidación con unas gotas de peróxido de hidrógeno al 33 %, a hidrólisis con hidróxido de sodio 1 mol/L y ácido clorhídrico 1 mol/L (1:1), colocándose las muestras durante 30 min en un baño de agua destilada a 100 °C. Previamente se analizó la sustancia de referencia y el placebo sin degradar y posteriormente fueron evaluadas por cromatografía líquida de alta resolución.9 Las muestras fueron diluidas con la fase móvil de manera que se obtuviera una concentración similar a 20 µg/mL.
Se comprobó la linealidad del sistema, preparando muestras a diferentes niveles de loratadina que representan el 68,100,128,148, 168 % de la concentración teórica del principio activo en el jarabe, evaluándose el coeficiente de correlación, la ecuación de la recta, la prueba de linealidad y la prueba de proporcionalidad.10
La repetibilidad se realizó con una muestra homogénea y común del lote 46 que representa el 100 % de la cantidad teórica declarada. La precisión intermedia se realizó con la misma muestra homogénea de concentración única, efectuándose las valoraciones en 2 equipos diferentes, 2 días diferentes; para su evaluación se realizó un análisis de varianza, a través de la determinación del coeficiente de variación, prueba de Fischer y de la t de Student.11
El estudio de exactitud se desarrolló por el método de recuperación, mediante la preparación de muestras con diferentes niveles de loratadina que representan el 80, 90, 110 y 120 % respectivamente de la concentración teórica del principio activo en el producto terminado, evaluándose los parámetros de la prueba de la t de Student, de Cochran, ecuación de la recta y coeficiente de correlación.10
Todos los resultados obtenidos fueron evaluados estadísticamente con la ayuda del programa Microcal Origin 5.0 para Windows.
Resultados
La figura muestra los resultados obtenidos en el estudio de especificidad del método. Como se observa en el cromatograma correspondiente a la muestra placebo no se obtuvo ninguna señal en la zona de interés, al ser comparado con la señal obtenida para la solución estándar de referencia y de la muestra, lo cual indica que los excipientes o sustancias auxiliares de la solución no interfieren en la determinación del principio activo. En cuanto a las muestras sometidas a condiciones drásticas de: hidrólisis básica, hidrólisis ácida y H2O2.
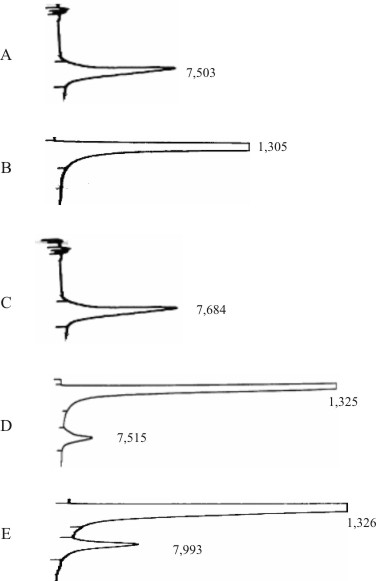
Fig. Estudio de especificidad. Cromatogramas: A) sustancia de referencia química;
B) placebo; C) muestra; D) muestra en medio ácido; E) muestra en medio básico.
Simultáneamente se realizó un scanning al pico correspondiente al principio activo en un rango de 200 a 500 nm, obteniéndose una pureza del 99,6 %.
La ecuación de la recta de la curva de calibración en el rango de concentraciones entre 13,6 a 33,6 µg/mL, se expresa según y = 0,9815 X + 0,0136, con un coeficiente de correlación lineal de 0,99854. Al aplicar la prueba de significación del intercepto se obtuvo una tcal = 3,91 para una ttab = 4,30. Cuando se aplicó la prueba de linealidad mediante los coeficientes de variación de los factores de respuesta CVf se obtuvo como resultado un coeficiente de variación CV= 2,1 %.
Los resultados del estudio de la precisión, realizado 2 días diferentes por 2 analistas diferentes, aparecen en la tabla 1.
Tabla 1. Resultados del estudio de la precisión
| Repetibilidad | Reproducibilidad | ||||
| Muestras | % | Analista 1 | Analista 2 | ||
| 1 | 98,0 | Día 1 | Día 2 | Día 1 | Día 2 |
| 2 | 98,1 | 100,4 % | 100,2 % | 97,8 % | 97,4 % |
| 3 | 98,5 | 97,8 % | 97,6 % | 98,3 % | 98,5 % |
| 4 | 97,9 | 98,5 % | 98,8 % | 99,1 % | 99,8 % |
| 5 | 98,3 | 97,6 % | 97,1 % | 98,5 % | 99,3 % |
| 6 | 98,2 | 98,6 % | 98,4 % | 98,5 % | 98,3 % |
| 7 | 98,5 | 98,3 % | 98,5 % | 98,6 % | 98,7 % |
| 8 | 99,0 | 98,2 % | 98,8 % | 98,4% | 98,5 % |
| 9 | 98,3 | Xm1 =98,5 % | Xm2 =98,5 % | Xm1 =98,5 % | Xm2 = 98,6 % |
| 10 | 99,1 | CV=0,9 % | CV= 0,9 % | CV= 0,4 % | CV =0,74 % |
| X media = 98,4 % DSR= 0,4 % CV= 0,41 % | Fcal =1,6 | tcal = 0,253 | |||
| Ftab (6/6; 0,05) = 4,28 | ttab (12; 0,05) = 2,18 | ||||
| Intervalo de confianza: 98,5 ± 0,04 % | |||||
En la tabla 2 se muestran los resultados obtenidos en el estudio de la exactitud del método. En la curva de recuperación, se reportan los mg teóricos contra los mg prácticos, obteniéndose la ecuación de la recta: y= 1,04885 X - 1,1107, con un coeficiente de correlación de 0,99624. Al aplicar la prueba de significación para conocer la influencia de la concentración (G de Cochran), se obtuvo una Gcal= 0,6923 para una Gtab= 0,8709. Al realizar la prueba de significación entre la recuperación media y el 100 % de recuperación, se obtuvo una tcal= 0,71 para una ttab= 2,45 .
Tabla 2. Resultados del estudio de exactitud
| mg adicionados | mg recuperados | Recobrado (%) | CV (%) | ||
| 0,01592 | 0,01591 | 0,01579 | 0,01579 | 98,6 | 0,50 |
| 0,01871 | 0,01825 | 0,01828 | 0,01822 | 101,8 | 1,25 |
| 0,02206 | 0,02218 | 0,02222 | 0,0222 | 100,52 | 0,023 |
| 0,02403 | 0,02411 | 0,02419 | 0,02418 | 100,32 | 0,023 |
Discusión
Los resultados obtenidos del análisis de la muestra placebo, la solución de referencia, la muestra del producto terminado y la muestra degradada por hidrólisis ácida, hidrólisis básica, oxidación H2O2, que se muestran en la figura, demuestran la especificidad del método al no presentarse interferencias de picos adicionales en la zona de elución del producto principal.6
El scanning realizado con el objetivo de determinar la pureza del pico de loratadina, en el rango de 200 a 500 nm demostró que no existía solapamiento de otros productos de degradación.
La curva de calibración resultó ser lineal en un rango de concentraciones entre 13,6 a 33,6 µg/mL.
Al aplicar la prueba del intercepto, este resultó ser no significativo ya que la tcal fue menor que la ttab para 4 g.l. y una probabilidad de 0,05. En la prueba de linealidad mediante los coeficientes de variación de los factores de respuesta CVf se obtuvo un coeficiente de variación menor del 5 %, lo que demuestra la adecuada linealidad de acuerdo con el límite establecido, por lo que se demuestra el cumplimiento de la linealidad en el intervalo de concentración estudiado.7
En el estudio de la repetibilidad se alcanzó un coeficiente de variación de 0,41 %, adecuado lo que nos demuestra la buena precisión del método, según el límite para los métodos cromatográficos CV £ 2 %.8 Los valores obtenidos para el estudio de la precisión intermedia demuestran que no existen diferencias significativas entre las precisiones alcanzadas por los analistas en diferentes días para una probabilidad de 0,05 % ya que el valor de Fcal es menor que la Ftab. Al realizar la prueba de la t de Student el valor calculado resultó ser menor que el tabulado para una probabilidad de 0,05 y 8 g.l., lo cual demuestra que no existen diferencias significativas entre las medias alcanzadas con un nivel de significación del 5 %.8
En el rango seleccionado en el estudio de exactitud, los valores de porcentaje de recobro estuvieron dentro de los límites establecidos para los métodos cromatográficos (98-102 %) y los valores del coeficiente de variación para cada uno de los niveles de concentración estudiados resultaron ser menor que el 2 %.11
En la influencia del factor concentración sobre la variabilidad de los resultados de la exactitud al aplicar la prueba de Cochran se obtuvo que Gexp fue menor que la Gtab para una probabilidad de 005, k= 3 y n= 3; por tanto, las varianzas de las concentraciones empleadas son equivalentes y nos indican que la concentración no influye en la variabilidad de estos.
No existen diferencias significativas entre la recuperación media (100,2 %) y el 100 % de recuperación al aplicar la prueba de la t de Student, ya que la tcal fue menor que la ttab para 6 g.l. y una probabilidad de 0,05, por lo que el método es exacto.
La curva de recuperación reportada de mg teóricos contra los mg prácticos demostró un comportamiento lineal, ya que la ecuación de la recta y el coeficiente de correlación se encontraron dentro de los límites establecidos para este ensayo. La prueba de significación del intercepto resultó no significativa y el valor de la pendiente no difiere significativamente de la unidad. Los valores alcanzados de la pendiente, el intercepto y el coeficiente de correlación demostraron la exactitud y linealidad del método.9
En conclusión, el método analítico desarrollado por CLAR para el estudio de estabilidad del jarabe de loratadina resultó ser específico, lineal, preciso y exacto en el intervalo de concentraciones de 13,6 y 33,6 µg/mL
Summary
Analytical method by high resolution liquid chromatography for the stability study of loratidine syrup 0.1 %
A high resolution liquid chromatography method was validated to study the stability of loratidine syrup 0.1 %. The calibration curve in the range from 13.6 to 3.36 µg/mL was lineal, with a coefficient of correlation equal to 0.99975. The intercept and slope statistical test was not significant. The recovery obtained was 100.2 % in the concentration range studied, and the Cochran and Student (t) tests results were not important. The variation coefficient in the repeatability study was equal to 0.41 % for 10 replications assayed, whereas in the reproducibility Fischer and Student tests were not remarkable. The method proved to be specific, lineal, accurate, and exact.
Key words: Loratidine, high resolution liquid chromatography, validation.
Referencias bibliográficas
- Goodman, Gilman, A. Las bases farmacológicas de la terapéutica. 3ra ed. Tomo II. La Habana: Edición Revolucionaria; 1994. p. 791.
- Martindale. The Extra Pharmacopoeia. 28 ed. London : The Pharmaceutical Press; 1989. p. 1248-9.
- Cromatografía en la Química Clínica. HPLC-Reactivos Merck. 1988. p. 3-16.
- Quattrocchi OA, Laba RF. Introducción a la HPLC. En Aplicación y práctica. Buenos Aires: Ed. Artes Gráficas Farro SA; 1992. p. 106-122, 284, 302-28.
- Castro M, Gascón S. Validación de métodos analíticos. Sección Catalana de AEFI. Comisión de normas de buena fabricación y control de la calidad. Sept, 1989. p. 1-94.
- United States Pharmacopoeial Convention. USP 29. Validation of compendial methods. 23 ed. Rockville : Mack Printing; 2006. p. 1982-4.
- International Conference on Harmonization (ICH) of Technical Requirements for the Registration of Pharmaceuticals for Human Use, Validation of analytical procedures, ICH-Q2A, Geneva, 1995.
- International Conference on Harmonization (ICH) of Technical Requirements for the Registration of Pharmaceuticals for Human Use, Validation of analytical procedures: Methodology, ICH-Q2B, Geneva, 1996. ICH harmonized tripartite guideline. Methodology of validation of analytical method. Recomendado en Nov. 1996.
- Sarrio RV, Silvestre LJ. Validation of Analytical Assays and Test Methods for the Pharmaceutical Laboratory. The Regulatory Forum. Bioserch. 1996.
- Guidance for Industry. Analytical Procedures and Methods Validation
Chemistry, Manufacturing, and Controls Documentation. FDA/Center for Drug Evaluation and Research. 2001. - Guidance for Industry. Analytical Procedure and Methods Validation. Draft Guidance. August, 2000.
Recibido: 11 de octubre de 2006. Aprobado: 17 de noviembre de 2006.
Lic. Leonid Torres Amaro Centro de Investigación y Desarrollo de Medicamentos (CIDEM). Ave. 26 No. 1605 entre Boyeros y Puentes Grandes, municipio Plaza de La Revolución. La Habana, CP 10 600, Cuba. Correo electrónico:E-mail: leonid@cidem.quimefa.cu
1 Licenciado en Ciencias Farmacéuticas. Investigador Agregado.
2 Master en Ciencias. Investigadora Agregada.

