










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Farmacia
versión On-line ISSN 1561-2988
Rev Cubana Farm v.42 n.3 Ciudad de la Habana sep.-dic. 2008
ARTÍCULOS ORIGINALES
Validación de un método espectrofotométrico para la cuantificación de principio activo en tabletas de nifedipino 10 mg
Validation of a spectrophotometric method for quantification of the active principle in 10 mg Nifedipine tablets
Yalexmi Ramos RodríguezI; Elsa Eneida Díaz ÁguilII; Alen Nils Baeza FonteI; Juan Luis Benítez CaballeroIII
ILicenciado en Ciencias Farmacéuticas. Laboratorios MedSol, Centro de Investigaciones del Ozono, Centro Nacional de Investigaciones Científicas. La Habana, Cuba.
IILicenciada en Química. Laboratorios MedSol, Centro de Investigaciones del Ozono, Centro Nacional de Investigaciones Científicas. La Habana, Cuba.
IIIMaster en Ciencias Química. Laboratorios MedSol, Centro de Investigaciones del Ozono, Centro Nacional de Investigaciones Científicas. La Habana, Cuba.
RESUMEN
Se realizó la validación de una técnica espectrofotométrica para la cuantificación del principio activo en tabletas de nifedipino 10 mg con el empleo de etanol al 96 % como solvente. Se determinaron los parámetros de linealidad, exactitud, precisión intermedia, repetibilidad, especificidad y robustez. Se comprobó que el método es lineal, exacto y preciso en un rango de 85 a 115 %, lo que demuestra la fiabilidad del método y su posibilidad de empleo en el control de la calidad de la forma terminada.
Palabras clave: Nifedipino, espectrofotometría UV, validación.
ABSTRACT
The validation of a spectrophotometric method for the determination of active principle in tablets of nifedipine 10 mg is presented in this work. The linearity, precision, exactitude, specificity and robustness of the method were evaluated. The method employed was linear, exact and precise in the interval of 85 % to 115%. This proves that the method can be used in the tablets' quality control.
Key words: Nifedipine, UV Spectrophotometric, Validation.
INTRODUCCIÓN
El nifedipino es un potente vasodilatador periférico que se usa en el tratamiento de la hipertensión arterial y en la angina de pecho crónica estable y la angina variante (de Prinzmetal).1-3
Muchos métodos se han reportado para la determinación de nifedipino, tanto en materia prima como en productos terminados. La espectrofotometría UV-Vis es uno de los métodos más utilizados para estos propósitos.4-7 La gran mayoría de los métodos reportados en la literatura permiten una cuantificación indirecta del nifedipino, ya que se requieren reactivos capaces de formar compuestos coloreados con el nifedipino para poder ser detectados luego en la zona visible del espectro.
En este trabajo se desarrolló un método analítico directo por espectrofotometría ultravioleta para la cuantificación de nifedipino en la forma terminada de tableta con en el empleo de etanol al 96 % como solvente.
El desarrollo de técnicas analíticas novedosas es un problema cotidiano. Siempre que esto sucede se exige como parte integral del estudio la validación del método en cuestión.8 La validación de un método es aquel proceso por el cual se establece mediante estudios de laboratorio que su capacidad satisface los requisitos para las aplicaciones deseadas.9 Este trabajo tiene como objetivos realizar la validación de una técnica espectrofotométrica para la determinación de nifedipino en la forma terminada tableta y proponer este método para su control químico de calidad.
MÉTODOS
Materiales
Las tabletas de nifedipino de lotes industriales así como el placebo correspondiente a dichas tabletas fueron suministradas por los Laboratorios MedSol, Cuba. Se empleó nifedipino estándar de referencia certificada en el Centro de Investigación y Desarrollo de Medicamentos (CIDEM) con una base hidratada (BH) de 99,51 %. Se utilizó papel toalla y papel de filtro cuantitativo.
Reactivos
Se utilizó etanol 96 % y metanol para análisis (Merck), etanol de producción nacional (cumple requerimientos USP-27) y patrones de productos de degradación dimetil 4-(2-nitrofenil)-2, 6-dimetilpiridina -3,5-dicarboxilato (nifedipino impureza A) y dimetil 4-(2-nitrosofenil)-2, 6-dimetilpiridina -3,5-dicarboxilato) (nifedipino impureza B) (USP).
Preparación de la disolución muestra de nifedipino
Se pesó 20 tabletas de nifedipino, se determinó el peso promedio y se trituraron en un mortero. Se pesó polvo de tabletas equivalente a 15 mg de nifedipino, se transfirió a un frasco volumétrico de 50 mL y se le adicionó 30 mL de etanol al 96 %, lo cual se agitó mecánicamente durante 30 min en una zaranda. Se enrasó con etanol al 96 % y se filtró, y se desecharon los primeros 15 mL del filtrado. Se pipetearon 5 mL y se transfirieron a un frasco volumétrico de 50 mL. Se enrasó con etanol al 96 %. Se obtuvo una solución con una concentración de 30 mg/L. Se determinó la absorbancia a 342 nm.
Preparación de la disolución de nifedipino patrón de referencia
Se pesaron 50 mg de nifedipino sustancia de referencia y se transfirió a un frasco volumétrico de 100 mL. Se adicionó 60 mL de etanol 96 % y se agitó hasta total disolución. Se enrasó con el mismo solvente, se pipetearon 3 mL de la solución, se transfirieron a un frasco volumétrico de 50 mL y se enrasó con etanol al 96 %. Se obtuvo una solución con una concentración de 30 mg/L. Se determinó la absorbancia a 342 nm. Una vez determinada la absorbancia de la muestra y el patrón de referencia, se calculó el porcentaje de principio activo mediante la ecuación siguiente:
Validación del método espectrofotométrico empleado en la cuantificación
En la validación del método se utilizaron como parámetros de aceptación los establecidos por la USP-27.9 Los parámetros estudiados fueron:
Linealidad: se prepararon 5 disoluciones muestras de nifedipino, cada una por triplicado, con concentraciones de 15; 21; 30; 37,5 y 45 µg/mL, correspondientes al 50, 70, 100, 125 y 150 % de la concentración de trabajo. Se construyeron curvas de absorbancia contra concentración y se efectuó un análisis de regresión, calculando el coeficiente de regresión (r), el coeficiente de determinación (r2), coeficiente de variación de los factores de respuesta (CVf), las pendientes, interceptos y significación de estas. Los criterios de aceptación son: CVf £ 5 %; r ³ 0,99; r2 ³ 0,98; intercepto no significativamente diferente de cero y la pendiente altamente significativa.
Exactitud: este ensayo se realizó por el método de los placebos cargados. Se prepararon disoluciones muestras de nifedipino con concentraciones de 25,5; 28,5; 30; 31,5 y 34,5 µg/mL correspondientes al 85, 95, 100, 105 y 115 % de la concentración de trabajo. Se compararon las medias de los porcentajes calculados con los porcentajes adicionados a través de una prueba de la t de Student así como las dispersiones para cada uno de los niveles evaluados mediante la prueba de Cochran. Se calcularon los estadígrafos totales expresados como porcentaje de recobro (% R) y coeficiente de variación (CV). Los criterios de aceptación son: % R = 97-103 %; CV £ 3,0 %. Se construyó una recta ploteando los porcentajes adicionados contra los porcentajes calculados. La similitud entre ambos parámetros se demuestra al obtener una recta con intercepto y pendiente no estadísticamente diferentes de 0 y 1 respectivamente, verificado a través de una prueba de la t de Student.
Precisión: este parámetro incluye la precisión intermedia y la repetibilidad.
Precisión intermedia: este ensayo se realizó con disoluciones muestras de nifedipino con concentraciones de 30 µg/mL correspondientes al 100 % de la concentración de trabajo, por cuadruplicado, por 2 analistas, durante 3 días sin variar las condiciones experimentales. Se compararon las dispersiones por días y por analistas a través de una prueba de G Cochran. El coeficiente de variación de los estadígrafos totales debe ser menor o igual al 3,0 %.
Repetibilidad: este ensayo se realizó con disoluciones muestras de nifedipino con concentraciones de 21; 30 y 34,5 µg/mL correspondientes al 70, 100 y 115 % de la concentra ción de trabajo, por el mismo analista, en el mismo laboratorio, el mismo día y con el mismo equipo y reactivos, realizando cada nivel por sextuplicado. Se compararon las dispersiones de los 3 niveles de concentración a través de una prueba de G Cochran. Los coeficientes de variación de los porcentajes de contenido para cada nivel de concentración deben ser menores o iguales al 1,5 %.
Especificidad al placebo: en este ensayo se compararon las regresiones lineales de soluciones de nifedipino patrón de referencia, correspondientes al 50, 70, 100, 125 y 150 % de la concentración de trabajo (concentraciones de 15; 21; 30; 37,5 y 45 µg/mL respectivamente), con placebo y sin este, no debió de existir diferencias significativas entre las pendientes y los interceptos de ambas rectas.
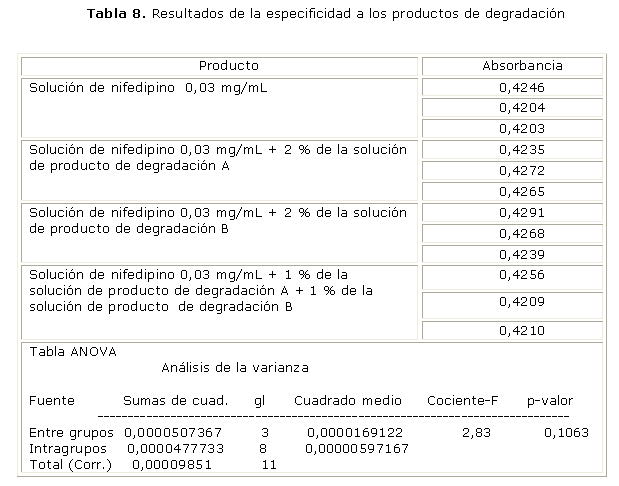
Especificidad a los productos de degradación: en este ensayo se prepararon 4 soluciones de trabajo: soluciones de nifedipino a la concentración de trabajo (30 µg/mL); soluciones de nifedipino con un 2 % del producto de degradación A; soluciones de nifedipino con un 2 % del producto de degradación B; y soluciones de nifedipino con un 2 % de la mezcla de ambos productos, cada solución preparada por triplicado. Se determinaron los espectros UV de soluciones de ambos productos de degradación para observar la posible interferencia de estos en la evaluación del principio activo. Se efectuó un análisis de varianza entre las muestras que contenían solamente al nifedipino y las que contenían el nifedipino más los productos de degradación; no debió existir diferencias significativas entre las absorbancias obtenidas entre las soluciones.
Robustez: en este ensayo se trabajó con porcentajes de etanol del 96; 94,9 y 92 %; papel de filtro cualitativo y papel toalla; longitudes de onda de lectura de 342 ± 3 nm y extracción del principio activo en zaranda y baño ultrasónico. Se evaluó cada parámetro estudiado la similitud o no de las poblaciones correspondientes al método normal propuesto y los alternativos.
El procesamiento estadístico de todos los ensayos del proceso de validación se realizó con el programa de cómputo STATGRAPHICS Plus versión 5.1.
RESULTADOS
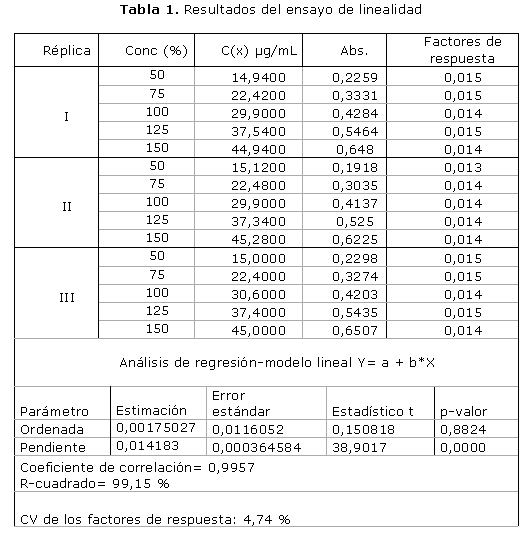
El primer parámetro evaluado en el proceso de validación fue la linealidad (tabla 1). El procesamiento estadístico de los resultados mostró valores de coeficientes de correlación r igual a 0,9957, coeficiente de determinación de 0,9915 y coeficientes de variación de los factores de respuesta de 4,74 %. El análisis estadístico de regresión ofreció un intercepto que no es estadísticamente diferente de cero: 0,00175 con una probabilidad de 0,8824. La pendiente resultó ser altamente significativa: 0,0142 con una probabilidad de 0.0000, o sea menor de 0,05.
En el parámetro de exactitud la comparación de las medias de los porcentajes calculados y los porcentajes adicionados, para cada nivel estudiado, mostró valores de p mayores de 0,05 (tabla 2). Se obtuvo un coeficiente de recobro total R de 100,13 % así como un CV de 0,49 %. En la tabla 3 se muestra el procesamiento estadístico de la recta obtenida al graficar % adicionado contra % recuperado. Para el caso del intercepto, este no difiere estadísticamente del valor cero dado por un p-valor superior a 0,05. De igual forma ocurre con la pendiente, al compararse con la población constituida por los factores de respuesta con el valor uno. En este caso se demostró que la pendiente no es diferente estadísticamente de uno para un 95 % de confianza dado por un p-valor de 0,187779, o sea superior a 0,05.

En el caso de la precisión intermedia (tabla 4), se obtuvieron valores de p asociado a la prueba de Cochran superiores a 0,05 (0,2798 entre días y 0,3665 entre analistas). El valor del coeficiente de variación resultó ser de 0.81%, o sea, menor del 3 %, límite superior establecido para este ensayo.
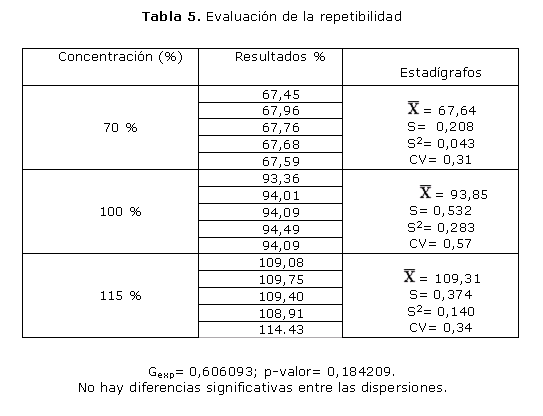
En la repetibilidad (tabla 5) se obtuvo un valor de p asociado a la prueba de Cochran de 0,1842, o sea, superior a 0,05. De igual forma se aprecia un valor total de CV de 0,41 %, o sea, menor de 1,5 %, límite establecido para este método.
En la especificidad del método al placebo se compararon las regresiones lineales obtenidas con placebo y sin este (tabla 6), se demostró que los interceptos no son diferentes significativamente de cero, dado por valores de p superiores a 0.05. La igualdad de las pendientes se evaluó mediante el método de contraste múltiple de rango (tabla 7) y se demostró que hay un 95 % de confianza de que ambas pendientes son iguales.
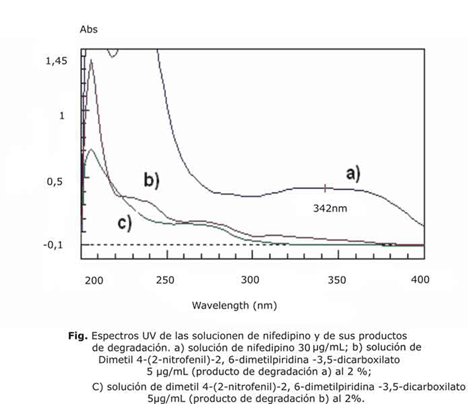
En la figura se observan los espectros UV de cada una de las especies en estudio para el caso de la especificidad a los productos de degradación. La contribución de absorbancia de cada uno de los productos a 342 nm aparentemente es limitada. Para determinar la magnitud de esta contribución se realizó un análisis de varianza entre las muestras que contenían solamente al nifedipino y las que contenían el nifedipino más los productos de degradación (tabla 8), se obtuvo un valor de p asociado a la prueba de Fischer de 0,1063, o sea, superior a 0,05.
En la tabla 9 se observan los resultados obtenidos en el parámetro robustez. Como se observa, para todas las condiciones se obtuvieron valores de p asociados a la prueba de Fisher superiores a 0,05.
DISCUSIÓN
Al analizar los resultados obtenidos en la linealidad se pudo demostrar que el valor de coeficiente de correlación r se acercó bastante a la unidad, además se obtuvo un coeficiente de determinación r2, coeficiente de variación de los factores de respuesta CVf, pendiente e intercepto dentro de los límites exigidos. Todo lo anterior permite afirmar que existe una buena correlación entre la concentración y la respuesta obtenida (valores de absorbancia), lo cual demuestra linealidad en el rango de concentraciones estudiadas.
En el ensayo de exactitud, los resultados se expresaron en función del coeficiente de recobro R y el coeficiente de variación CV. Como se puede evidenciar en la tabla 2, el método propuesto es exacto, ya que los valores de porcentajes de recobro y coeficientes de variación se encuentran dentro del intervalo establecido. El ploteo de % adicionado contra % recuperado mostró una recta con intercepto y pendiente no estadísticamente diferentes de cero y uno respectivamente Este resultado demuestra que nos encontramos frente a una recta tipo y= x, por lo que se puede afirmar que el procedimiento no tiene sesgo ya que se logra la recuperación total del nifedipino.
Los resultados del estudio de precisión intermedia indican que no existen diferencias estadísticamente significativas entre las desviaciones estándares de los resultados obtenidos en los 3 días ensayados y por los 2 analistas, dado por valores de probabilidad (entre los días y entre los analistas) mayores de 0,05 para un 95 % de confiabilidad. Se obtuvo un valor de CV menor del límite establecido para este método, lo que demuestra que los resultados son independientes del día en que se haga el análisis y de la habilidad del analista que lo ejecute.
En el ensayo de repetibilidad se analizaron los niveles de concentración equivalentes al 70, 100 y 115 % de la concentración de trabajo. Se considera bajo el 70 % ya que el límite de aptitud del producto terminado es entre 90 y 110 % de la cantidad establecida. El valor de 115 % se considera alto pues constituye el límite superior establecido para la uniformidad de contenido. Cuando se compararon las dispersiones para los 3 niveles de concentraciones estudiadas, se demostró la no existencia de diferencias estadísticamente significativas ya que el valor de p asociado a la prueba de Cochran resultó ser mayor de 0,05 para un 95 % de confianza. El valor total de CV fue menor del 1,5 %, límite establecido para este método. Estos resultados nos permiten afirmar que el método es repetible en los 3 niveles de concentración estudiados, por cumplir los parámetros establecidos como límite de aceptación.
Los resultados de la especificidad del método al placebo demuestran que las sustancias auxiliares (placebo) no influyen en la respuesta analítica del nifedipino.
En el caso de la especificidad del método a los productos de degradación se demostró la no existencia de diferencias significativas entre las lecturas obtenidas para la solución de nifedipino y las que contienen productos de degradación, ya que se obtuvo un valor de p superior a 0,05 para un 95 % de confianza. Esto confirma que los productos de degradación no interfieren en la determinación del nifedipino. Es importante mencionar que la técnica objeto de validación cuenta con un ensayo de sustancias relacionadas por cromatografía de placa delgada, lo que permite solo un contenido de los productos de degradación de 0,1 %.
Los resultados obtenidos en el ensayo de robustez demuestran la no existencia de diferencias estadísticamente significativas para cada uno de los parámetros estudiados para un 95 % de confianza. Esto permite afirmar que el método es robusto en las condiciones de estudio.
En conclusión, el método propuesto es lineal, preciso y exacto en el rango del 85 al 115 % de la concentración de trabajo; también es específico, se probó la no interferencia del placebo ni de los productos de degradación del principio activo; los resultados obtenidos prueban la fiabilidad del método para la cuantificación del principio activo en tabletas de nifedipino.
REFERENCIAS BIBLIOGRÁFICAS
1. Martindale. The Extra Pharmacopoeia. 33 ed. London: Royal Pharmaceutical Society; 2002. p. 940-6.
2. Alfonso R. Gennaro. Remington's Pharmaceutical Sciences. 18 ed. Pennsylvania: Marck Publishing Company; 1990. p. 855.
3. Diccionario de Especialidades Farmacéuticas. 49 ed. México, DF: Adalat; 2003.
4. Bruno L, John SK. New spectrophotometric methods for estimation of nifedipine. Indian J Pharmaceut Sci. 1988;50(2):109-12.
5. Rahman N, Hejaz Azmi SN. New spectrophotometric methods for the determination of nifedipine in pharmaceutical formulations. Il Farmaco. 2005;52(4):915-22.
6. Rahman N, Ahmad Khan N, Hejaz Azmi SN. Extractive spectrophotometric methods for the determination of nifedipine in pharmaceutical formulations using bromocresol green, bromophenol blue, bromothymol blue and eriochrome black T. Il Farmaco. 2004;59(1):47-54.
7. Rahman N, Hoda MN. Spectrophotometric method for the determination of nifedipine with 4-(methyl amino) phenol and potassium dichromate. Il Fármaco. 2002;57(6):435-41.
8. Díaz de Armas M, Hernández Oramas I, Martínez Cuervo M. Validación de técnicas analíticas utilizadas en el control de calidad. Rev Cubana Farm. 1998;32(2):1.
9. United States Pharmacopoeia Convention. Farmacopea de los Estados Unidos USP. Validation of Compendial Methods. 27 ed. Rockville: Maryland; 2004.
Recibido: 25 de abril de 2008.
Aprobado: 2 de junio de 2008.
Lic. Yalexmi Ramos Rodríguez. Laboratorios MedSol. Centro de Investigaciones del Ozono, Centro Nacional de Investigaciones Científicas. Código Postal 6 412, La Habana, Cuba. Correo electrónico: yalexmi.ramos@cnic.edu.cu


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}