










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Farmacia
versión impresa ISSN 0034-7515versión On-line ISSN 1561-2988
Rev Cubana Farm v.43 n.2 Ciudad de la Habana Mayo-ago. 2009
ARTÍCULOS ORIGINALES
Desarrollo de la formulación de la suspensión oral de ibuprofeno 100 mg/5 mL para uso pediátrico
Development of formulation for oral suspension Ibuprofen 100 mg/5mL for pediatric use
Nilia de la Paz Martín-ViañaI; Iván Gastón Morales LacarrereII; José Manuel Gil ApánIII; Dunia Casanave GuanaluceIV; Pedro Barzaga FernándezIV; Ramona Núñez GomeroIII
ILicenciada en Ciencias Farmacéuticas. Máster en Ciencias Tecnología y Control de Medicamentos. Investigadora Auxiliar. Centro de Investigación y Desarrollo de Medicamentos. (CIDEM). La Habana, Cuba.
IILicenciado en Ciencias Farmacéuticas. Investigador Agregado. CIDEM. La Habana, Cuba.
IIITécnico Medio en Química. CIDEM. La Habana, Cuba.
IVLicenciado en Ciencias Farmacéuticas. Aspirante a Investigador. CIDEM. La Habana, Cuba.
RESUMEN
Se empleó el método de prueba y error para el desarrollo de la formulación de la suspensión oral de ibuprofeno 100 mg/5 mL para uso pediátrico; se estudió su estabilidad químico-física por el método acelerado y de vida de estante; se envasó en frasco de vidrio ámbar por 120 mL y se almacenó a temperatura ambiente. Se realizó el estudio reológico y la determinación de la viscosidad aparente, además, se efectuó el estudio microbiológico a través de la prueba de efectividad de preservativo antimicrobiano y el conteo microbiano; se comprobó la seguridad del producto mediante el estudio toxicológico. Todos los resultados cumplieron con los límites de calidad establecidos en la literatura científica, USP 30, para este tipo de forma farmacéutica. Se concluye que el medicamento desarrollado está correctamente formulado, desde el punto de vista galénico con un tiempo de vida útil de 24 meses bajo las condiciones estudiadas.
Palabras clave: Estabilidad, ibuprofeno, suspensión.
ABSTRACT
Authors used the test and error method to develop the formulation of Ibuprofen oral suspension (100 mg/5 mL) for pediatric use. Its chemical-physic stability was studied through accelerated method and shelf life. It was bottled in amber glass small bottles by 120 mL, and it was stored at room temperature. A rheology study and assessment of apparent viscosity was made as well as a microbiologic one by test of effectiveness of antimicrobial preservative and the microbial count. Product safe was verified by toxicology study. All results fulfilled quality limits established in scientific literature, USP 30, for this type of pharmaceutical method. We conclude that drug developed is correctly formulated, from the doctoral point of view with a time of useful life of 24 months under study conditions.
Key words: Stability, Ibuprofen, suspension.
INTRODUCCIÓN
El ibuprofeno, derivado del ácido propiónico, es una mezcla racémica de enantiómeros R y S con acción antiinflamatoria no esteroidal, analgésica y antipirética. Este fármaco se administra por vía oral y se comercializa en la actualidad en varias formas farmacéuticas (suspensión, grageas, tabletas convencionales y de liberación controlada). Este medicamento fue aprobado para su uso y distribución en el año 1974 por la Food and Drug Administration.1-7
En el presente trabajo se propuso desarrollar una suspensión de ibuprofeno 100 mg/5 mL, para ser administrada a la población pediátrica, por vía oral, que cumpliera con los índices de control de calidad para esta forma farmacéutica y así obtener una formulación segura, efectiva y eficaz para el paciente.
Por tanto, el objetivo fundamental consistió en desarrollar una suspensión oral de ibuprofeno que brindara una dosis de 100 mg por cada 5 mL de preparación, que cumpliera con todos los requerimientos para esta forma farmacéutica y que proporcionara el efecto terapéutico deseado. Como objetivos específicos para lograr lo anteriormente planteado, se propuso desarrollar la formulación por el método de prueba y error, previa comprobación de la calidad farmacéutica tanto del principio activo como de las sustancias auxiliares utilizadas en su composición, estudiar su estabilidad químico-física por el método acelerado y de vida de estante, mediante el empleo de la técnica analítica descrita en la United States Pharmacopeial 308 (USP 30), realizar el estudio del comportamiento reológico y determinar la viscosidad aparente con la utilización de un rotoviscosímetro de la firma HAAKE. Además, realizar el estudio microbiológico a través de la prueba de efectividad de preservativo antimicrobiano y el conteo microbiano al inicio y al final del estudio según se describe en la USP 309 y comprobar la seguridad del producto mediante el estudio toxicológico.
MÉTODOS
Se desarrolló la formulación con el empleo el método de dispersión, para ello se empleó como principio activo el ibuprofeno (Hubel, China) y sustancias auxiliares de uso común en la Industria Farmacéutica para la elaboración de este medicamento; en la tabla 1 se muestra su composición cualitativa.
Se elaboraron 3 lotes pilotos, correspondientes a la formulación seleccionada; la suspensión se envasó en frascos de vidrio ámbar, por 125 mL de capacidad nominal, con tapa para frascos pilfer. Se almacenaron a temperatura ambiente (30 ± 2 °C), y se rotularon como lote 03001, 03002 y 03003.
Una vez elaborados los 3 lotes se evaluaron las características organolépticas, se determinó el pH, la disolución y la valoración del fármaco en el medicamento, como control de calidad, sigún la monografía descrita en la USP 30.8
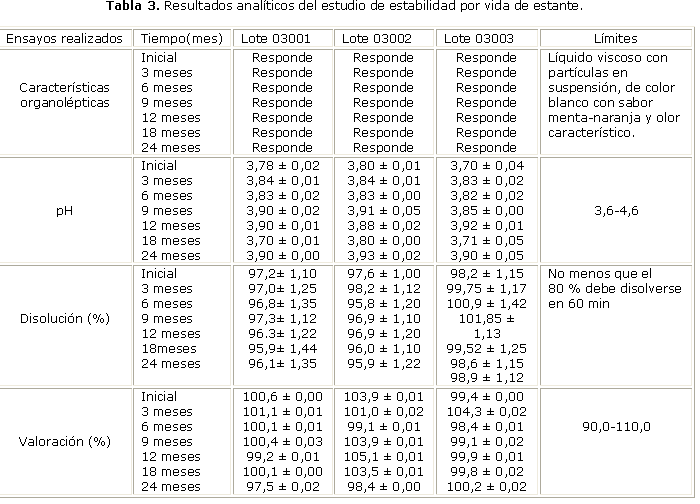
El estudio de estabilidad química se efectuó por el método acelerado y de vida de estante. El estudio de estabilidad acelerado se realizó colocando muestras de los lotes 03001, 03002 y 03003 a 40 ± 2 °C y 75 ± 5 % de humedad relativa (HR), las cuales se analizaron al inicio, 1, 2, 3 y 6 meses de elaborados. Para el estudio de estabilidad por vida de estante las muestras fueron almacenadas a temperatura ambiente (30 ± 2 °C) y protegidas de la luz, las que fueron evaluadas al inicio, a los 3, 6, 9, 12, 18 y 24 meses de fabricados.
Se empleó para la suspensión de ibuprofeno el método analítico descrito en la USP 30;8 se utilizó como fase móvil una mezcla de la solución de ácido fosfórico 0,01 M y acetonitrilo (63:37); la columna empleada fue RP-8 de 150 x 4 mm y 5 µm a una longitud de onda de 220 µm con un loop de 20 µL.
Para el estudió del comportamiento reológico y de la viscosidad aparente, se empleó un rotoviscosímetro Haake, modelo RV 20, con sistema sensor M5/NV y un gradiente de velocidad comprendido entre 0-50 s-1 por un tiempo de 2 min. Se trabajó en ambiente termostatado a 30,0 ± 0,1 °C.
Se realizó el análisis estadístico de los resultados del estudio de viscosidad aparente, al inicio y decursados 12 y 24 meses, a través de un ANOVA factorial, clasificación doble efecto fijo, previa la comprobación de la homogeneidad de varianza, con el empleo de la prueba de Barlett y por último se efectúo el ensayo de Duncan.
Se efectuó la prueba de efectividad de preservativos antimicrobianos, según USP 30.9 Las muestras fueron contaminadas con una batería de microorganismos compuesta por: S. aureus, P. aeruginosa, E. coli, C. albicans y A. níger, en una concentración de 500 000 a 1 000 000 de m.o./mL. Se realizaron siembras inmediatamente después de la contaminación del producto en un medio apropiado y se determinó la concentración de microorganismos a los 7, 14, 21 y 28 d. A la preparación se le realizó el conteo de microorganismos totales al inicio y final del estudio, según USP 30.9
La determinación de la toxicidad aguda del medicamento, se realizó de acuerdo con las regulaciones internacionales establecidas10 y las Normas Éticas fijadas por el Consejo de Organizaciones Internacionales de Ciencias Médicas11 para el manejo de animales en la experimentación. Se emplearon 24 ratas Wistar (12 hembras y 12 machos) de peso corporal entre 150 y 200 g, procedentes de la colonia de la UCTB. Control Biológico, CIDEM, las cuales fueron mantenidas en un cuarto a temperatura controlada de 20 ± 2 °C, ciclos de luz/oscuridad de 12-12 h y humedad relativa entre 40 y 75 %. La alimentación consistió en ratonina paletizada (CENPALAB, La Habana) y agua a voluntad.
Se confeccionaron 4 grupos de 3 animales cada uno: placebo (hembras), placebo (machos), suspensión, lote 03001 (hembras) y suspensión, lote 03001 (machos). Los animales fueron identificados individualmente para la dosificación exacta de acuerdo con su peso corporal mediante un sistema de marcaje con ácido pícrico.
Para evaluar el potencial tóxico de la sustancia de ensayo se procedió a realizar el ensayo de clase tóxica aguda (CTA) regulado por la OECD en su norma No. 423.12, 13
La administración de las sustancias se realizó, directamente, desde sus frascos por vía oral mediante cánula intragástrica, para lo cual se les retiró la comida a los animales 16 h antes de su suministro.
En un inicio se administró de forma aguda la sustancia de prueba (200 mg/kg) y el placebo, ambos con un factor de volumen de 0,01 mL/g. Se comenzó con esta dosis a partir de datos que referían una DL50 de 1 050 mg/kg para ratas por vía oral.13 En una segunda experiencia se administró una dosis superior a 800 mg/kg de la sustancia de ensayo y el placebo, para ello se realizaron 2 administraciones espaciadas en un tiempo de 4 h, utilizando en cada una de ellas un factor de volumen de 0,02 mL/g. Por razones de volumen no se pudo administrar la dosis de 2 000 mg/kg. En ambas experiencias se trabajó con los grupos de hembras y posteriormente con los machos.
Los animales fueron observados constantemente durante las primeras 24 h, se continuó esta directamente durante un período de 14 d, y se registró cualquier síntoma tóxico. Al finalizar este período se procedió al sacrificio por inhalación de éter dietílico para realizarles la autopsia; se efectuó un examen macroscópico minucioso de órganos y tejidos. El peso corporal se registró al inicio, a los 7 d y al final del experimento.
RESULTADOS
Se obtuvo una formulación líquida en forma de suspensión, de color blanco, con sabor y olor característico (menta-naranja).
En la tabla 1, se muestran los valores de los diferentes ensayos realizados para el control de calidad del medicamento según los límites establecidos en cada caso, así como los del estudio de estabilidad química-física por el método acelerado y de vida de estante (tablas 2 y 3). Los valores de viscosidad aparente (tabla 4), y su análisis estadístico responden a un fluido con adecuada viscosidad, característica de una suspensión correctamente formulada.
Tabla 4. Resultados de la viscosidad aparente (mPa∙s)
| Lote | Inicio | 12 meses | 24 meses |
| 03001 | 371,3 | 236,3 | 204,4 |
| 03002 | 340,5 | 208,0 | 198,3 |
| 03003 | 325,5 | 179,7 | 195,3 |
| Análisis estadístico: nivel de significación 1 % | |||
| Homogeneidad de varianza | |||
| Barlett: 2,368 | |||
| ANOVA factorial | |||
| Fuente de variación | Grados de libertad | F | |
| Tratamientos | 8 | 66,28 | |
| Prueba de Duncan/lotes | |||
| Medias ordenadas de forma descendente/lote | Tamaños muestrales | ||
| 256,7/03001 | 9 | ||
| 244,4/03002 | 9 | ||
| 237,8/03003 | 9 | ||
| Lote | Diferencia | Resultado estadístico | |
| 03001 vs. 03002 | 12,2 | No significativo | |
| 03001 vs. 03003 | 18,8 | No significativo | |
| Prueba de Duncan/tiempos | |||
| Medias ordenadas de forma descendente/tiempo | Tamaños muestrales | ||
| 343,2/inicio | 9 | ||
| 208,0/6 meses | 9 | ||
| 187,7/12 meses | 9 | ||
| Tiempo | Diferencia | Resultado estadístico | |
| Inicio vs. 12 meses | 135,778 | Significativo | |
| 12 meses. 24 meses | 20,2777 | No significativo | |
Al inicio y decursados 24 meses se obtuvieron los reogramas correspondientes a un cuerpo del tipo no newtoniano; en la figura 1 se muestra un flujo seudoplástico con tixotropía, tal comportamiento reológico se mantuvo durante el tiempo de estudio y fue similar en los 3 lotes.

En la efectividad de preservativo antimicrobiano germinaron en la primera siembra, 100 m.o./mL de E. coli, 10 000 m.o./mL de P. aeruginosa, C. albicans, A. níger y 1 000 m.o./mL de B. subtilis. A los 7 y 14 d germinó 100 m.o./mL de B. subtilis. A los 21 y 28 d no se detectó contaminación.
Los conteos microbiológicos, al inicio y decursados 24 meses, mostraron un conteo de bacterias menor de una unidad formadora de colonia por mL (UFC/mL) y menor de 10 UFC/mL para el caso de los hongos.
No se observaron síntomas tóxicos a las dosis administradas, tampoco se presentó mortalidad en el período de observación, ni decremento en el peso corporal en ninguno de los grupos experimentales durante el estudio. En la tabla 5 se presentan los resultados del comportamiento del peso corporal durante el estudio para hembras y machos, respectivamente. En la autopsia realizada a los animales no se encontraron evidencias de alteraciones patológicas en los órganos analizados.
DISCUSIÓN
La formulación desarrollada cumplió con los requerimientos de calidad farmacéutica establecidos, para este producto en la USP 30,8 por lo que se obtuvo un medicamento correctamente formulado. Los tres lotes elaborados resultaron satisfactorios al cumplir con los límites establecidos en la literatura oficial.8
Al analizar los resultados presentados en las tablas 2 y 3 es evidente que en todos los estudios efectuados se han obtenido resultados satisfactorios, lo cual es indicativo de una formulación químicamente estable durante los 24 meses en observación y análisis, bajo las condiciones de envase y almacenamiento estudiadas.
Al evaluar los resultados y el análisis estadístico de los valores de la viscosidad aparente se observó que existen diferencias estadísticas, con un nivel de significación de un 1 %, entre los valores iniciales y los obtenidos a los restantes tiempo de estudio, no así entre los determinados a los 12 y 24 meses donde no se obtienen diferencias estadísticas, con una confiabilidad de un 99 %. Esta disminución de la viscosidad aparente, lo cual también se puede observar al analizar las curvas de viscosidad y fluidez (fig. 1 y 2), respectivamente, es característica de las preparaciones que presentan carboximetilcelulosa sódica en su composición, lo que no afecta la redispersabilidad de la suspensión, ni sus características mecánico-estructurales, porque no se observa formación de una torta dura o cake y al agitar la preparación se redispersa en un tiempo menor a 3 min y permanece homogénea por un período mayor de 48 h, este comportamiento fue observado en los 3 lotes.

Al inicio y decursados 24 meses se obtuvieron los reogramas correspondientes a un cuerpo del tipo no newtoniano, pues como se puede observar, la gráfica no constituye una línea recta que parte del origen de coordenadas (fig. 1). La figura 2 muestra un flujo seudoplástico con tixotropía, a medida que aumenta el gradiente de velocidad (D) y el valor del esfuerzo cortante (r), ocurre una pérdida de estructura del sistema, donde las partículas se alinean y la suspensión comienza a fluir sin necesidad de un esfuerzo cortante inicial, recuperando parte de su estructura y viscosidad inicial, cuando transcurrió un período de tiempo determinado al disminuir el gradiente de velocidad y el esfuerzo cortante, de manera que se mantiene estable su comportamiento reológico.
De acuerdo con los resultados del estudio de efectividad de preservativo antimicrobiano, se llega al criterio que el preservativo antimicrobiano empleado protege a la preparación de la contaminación microbiana, porque concluido el tiempo de estudio no se observan ninguno de los microorganismos con los que se contaminó la preparación.
Los resultados del conteo microbiano al inicio y decursados 24 meses cumplieron con el límite establecido, en la USP 30,9 al no existir microorganismos patógenos y el conteo de bacterias y de hongos ser menor de 103 UFC/mL y 102 UFC/mL, respectivamente.
Al analizar estadísticamente la ganancia de peso corporal mediante un análisis de varianza de una vía se observó que no existían diferencias significativas, con una confiabilidad de un 95 %, entre los grupos tratados y placebo en ambos sexos.
El ibuprofeno suspensión oral no presentó toxicidad significativa, con una confiabilidad de un 95 %, tras su administración oral a una dosis de 800 mg/kg, con el empleo de un factor de 0,02 mL/g. Este factor se considera límite para la especie y vía de administración empleadas, por lo que no fue posible la administración de la dosis de 2 000 mg/kg además el valor de DL50 reportado es de 636 mg/kg.14
Se demostró que este medicamento está correctamente formulado desde el punto de vista galénico, que cumple con los requisitos físico-químicos, microbiológicos y toxicológicos exigidos para una suspensión farmacéutica de uso en humanos, y resulta estable por un período de 24 meses, bajo las condiciones de almacenamiento y envase en que ha sido ensayado.
REFERENCIAS BIBLIOGRÁFICAS
1. Martindale. The complete drug reference. 32 ed. London: Staff; 2000. p. 90.
2. Medical Economics Company PDR. 54 ed. New York: Medical Economics Company; 2000. p. 2218-20.
3. Index Merck. 11 ed. New Jersey: Merck; 1989. p. 776.
4. A. Wolters Kluwer Company. Drugs Facts and Comparisons. 54 ed. St Louis: Facts and Comparisons; 2000. p. 817-8.
5. Diccionario de Especialidades Farmacéuticas. 47 ed. Lima: Ediciones PLM; 2001. p. 1715-6.
6. Diccionario de Especialidades Farmacéuticas. Santafé de Bogotá: PLM; 1991. p. 878-9, 1108-11, 1190-1.
7. Consejo General de Colegios Oficiales de Farmacéuticos. Catálogo Especialidades Farmacéuticas. Madrid: Consejo General de Colegios Oficiales de Farmacéuticos; 2000. p. 1577-9.
8. Farmacopea de los Estados Unidos de América. Formulario Nacional: Ibuprofeno suspensión oral. Rockville: Port City Press; 2007. p. 2602-3.
9. Farmacopea de los Estados Unidos de América. Formulario Nacional. Pruebas microbiológicas. Rockville: Port City Press; 2007. p. 85-100.
10. Guide for the care and use of laboratory animals. National Research Council. Washington: National Academy Press; 1996. p. 21-79.
11. Consejo de Organizaciones Internacionales de Ciencias Médicas. Principios Guías Internacionales para la Investigaciones Biomédicas que impliquen animales. Normas, documentos y ética médica. Ginebra, 1985. p. 97-107.
12. Testing of Chemical. Acute Oral Toxicity_Acute Toxic Class Method Nº 423 Adopted 20 December 2001.
13. Commission of the European Communities. Annex to Commission Directive 92/69/EEC of 31 July 1992 adapting to technical progress for the seventeenth time Council Directive 67/548/EEC on the approximation of laws, regulations and administrative provisions relating to the classification, packaging and labeling of dangerous substances. B1. Acute toxicity (oral). J Eur Comm. (L 383a). 1992;5:110-12.
14. Adams SS, Bough RG, Cliffe EE, Lessel B, Mills RFN. Absorption, distribution and toxicity of ibuprofen. Toxicol Appl Pharmacol. 1969; 15(2).
Recibido: 12 de enero de 2009.
Aprobado: 17 de febrero de 2009.
Lic. Nilia de la Paz Martín-Viaña. Centro de Investigación y Desarrollo de Medicamentos. (CIDEM). Calle 19 de Mayo No. 13, esquina a Amézaga, municipio Plaza de la Revolución, CP 10 600. La Habana, Cuba.


{kind=link}
{kind=link}
{kind=link}
{kind=link}