










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Farmacia
versión On-line ISSN 1561-2988
Rev Cubana Farm v.43 n.2 Ciudad de la Habana Mayo-ago. 2009
ARTÍCULOS ORIGINALES
Validación de un método cromatográfico para la cuantificación de mefenesina en tabletas de producción nacional
A chromatographic method validation to quantify tablets Mephenesine of national production
Yania Suárez PérezI; Adalberto Izquierdo CastroII; Jana María Milián SánchezIII
IDoctora en Ciencias Farmacéuticas. Licenciada en Ciencias Farmacéuticas. Máster en Ciencias Tecnología y Control de Medicamentos. Profesora Auxiliar. Instituto de Farmacia y Alimentos. Universidad de La Habana. La Habana, Cuba.
IILicenciado en Ciencias Farmacéuticas. Máster en Ciencias Tecnología y Control de Medicamentos. Instituto de Farmacia y Alimentos. Universidad de La Habana. La Habana, Cuba.
IIILicenciada en Ciencias Farmacéuticas. Instituto de Farmacia y Alimentos. Universidad de La Habana. La Habana, Cuba.
RESUMEN
Se realizó la validación de un método analítico por cromatografía líquida de alta eficiencia para la cuantificación de mefenesina en tabletas de 500 mg reformuladas recientemente. Con respecto a su aplicación al control de calidad, la validación incluyó los parámetros linealidad, exactitud, precisión y selectividad. Los resultados fueron satisfactorios en el rango de 50-150 %. Para su empleo en estudios posteriores de estabilidad química, se evaluó adicionalmente la selectividad para estabilidad y la sensibilidad. Los límites de detección y cuantificación estimados resultaron adecuados y el método fue selectivo frente a los posibles productos de degradación.
Palabras clave: Mefenesina, estabilidad, cromatografía líquida de alta eficiencia, tabletas, control de calidad.
ABSTRACT
Authors made validation of an analytical method by high performance liquid chromatography (HPLC) for quantification of Mephenesine in recently reformulated 500 mg tablets. With regard to its application to quality control, validation included the following parameters: linearity, accuracy, precision, and selectivity. Results were satisfactory within 50-150 % rank. In the case of its use in subsequent studies of chemical stability, the selectivity for stability and sensitivity was assessed. Estimated detection and quantification limits were appropriate, and the method was selective versus the possible degradation products.
Key words: Mephenesine, stability, high performance liquid chromatography, tablets, quality control.
INTRODUCCIÓN
La mefenesina es un relajante muscular y sedativo moderado, que apreció en el mercado en 1947. Tiene acción selectiva sobre el sistema nervioso central y se indica en enfermedades que provoquen hipertonía muscular, extrapiramidales, entre otras.I Se formula en tabletas de 500 mg, en cremas al 2 % y en ungüentos al 10 %.2
En Cuba se introduce en los Laboratorios NOVATEC, en forma de tabletas con 500 mg de dosis, las cuales ha sido recientemente reformuladas.3 Como parte de los estudios requeridos, se encuentra la validación de los métodos analíticos. Estos incluyen los métodos destinados a cuantificar el fármaco en las tabletas recién elaboradas, es decir, al control de calidad. Además se encuentran aquellos métodos necesarios para dar seguimiento a la formulación desde el punto de vista de la estabilidad química. Estos estudios son de vital importancia y deben realizarse según las exigencias actuales.4
MÉTODOS
Descripción del método por CLAE para el análisis de mefenesina en tabletas
El cromatógrafo empleado estaba constituido por:
- Bomba K-1001.
- Inyector manual con loop de 20 µL.
- Detector UV variable K-2500 acoplado a una computadora para la adquisición de los cromatogramas mediante el software CHROMGATE (versión 3.1).
Las condiciones cromatográficas establecidas fueron las siguientes:
- Fase móvil: acetonitrilo-agua (400:600).
- Fase estacionaria: columna LiChrosorb RP-18 25 cm x 4,6 mm 5 µm.
- Longitud de onda (l): 278 nm.
- Detector: UV.
- Flujo: 1mL/min.
- Temperatura ambiente ( 30 ± 0,5 °C).
Preparación de la solución muestra
1. Pesar individualmente 20 tabletas de mefenesina (lote 25D7) y determinar el peso promedio.
2. Triturar las tabletas en un mortero hasta obtener un polvo fino.
3. Pesar el polvo equivalente a 100 mg de principio activo y trasvasarlo a un matraz aforado de 100 mL.
4. Añadir 50 mL de fase móvil y colocarlo en el baño ultrasónico hasta total disolución.
5. Completar a volumen con fase móvil.
6. Homogeneizar.
7. Filtrar la muestra por papel de filtración rápida y desechar los primeros mililitros del filtrado.
8. Tomar una alícuota de 1,0 mL y trasvasar a un matraz aforado de 10 mL.
9. Completar a volumen con la fase móvil
Luego de preparar la solución a la concentración deseada, se inyectaron al cromatógrafo 20 µL para cada análisis realizado de solución muestra y de solución de referencia (0,1 mg/mL). Posteriormente se registraron los cromatogramas y se obtuvieron los valores del área bajo la curva (AUC) correspondientes.
Procedimiento aplicado para el cálculo
La concentración de la muestra (Cm) (mg/mL) se determinó por comparación con la respuesta analítica que se obtuvo para la solución de referencia (SR) de la cual se conocía su concentración. La expresión utilizada fue la siguiente:
Cm= (Cp x Am) / Ap
donde:
Cp: concentración del patrón (SR) expresada en %.
Am: Área bajo la curva (AUC) del pico obtenido para la muestra.
Ap: Área bajo la curva (AUC) del pico obtenido para el patrón (SR).
Validación del método analítico para el control de calidad
Se realizó la validación del método para el control de calidad teniendo en cuenta los parámetros exigidos para la categoría I según USP 28, 2005.5
Linealidad del sistema: Se realizó mediante el análisis de 5 concentraciones de mefenesina materia prima por triplicado, en un rango de 50-150 % de la cantidad teórica declarada como 100 % (1,0 mg/mL), equivalentes a 50, 75, 100, 125, 150 µg/mL respectivamente.
Se construyó una curva de calibración de área bajo la curva (AUC) vs. concentración teórica (%). Los resultados se procesaron estadísticamente a través del paquete STATISTICA for Windows versión 6.01 (opción regresión lineal múltiple) y se determinó: r (coeficiente de correlación lineal), r2 (coeficiente de determinación), a (intercepto) y b (pendiente), para el 95 % de confianza.
Criterios de aceptación
- Ecuación de la recta: y= bx + a
- r ³ 0,99
- r2 ³ 0,98
- Prueba de proporcionalidad del método analítico o hipótesis nula de la ordenada en el origen a= 0
- Se empleó la prueba estadística t de Student para n-2 grados de libertad, siendo n el número total de valores donde: texp < ttab.
- Prueba de la hipótesis nula de la pendiente: b= 0. Se determinó a partir de una prueba ANOVA de la regresión, teniendo en cuenta la probabilidad asociada al valor de la pendiente, es decir, si la p < 0,05, el valor de "b" difiere significativamente de cero.
- Se calcularon los factores de respuesta (ƒ) según la expresión:
![]()
donde:
y: respuesta analítica (AUC)
x: cantidad de analito (%)
Con estos resultados se determinó el valor medio, la DE y el coeficiente de variabilidad (CV). El CV debe ser menor que 5 %.
Linealidad del método: Este parámetro se evaluó en un rango de 50-150 %, preparando placebos cargados con 5 concentraciones crecientes del principio activo; se analizaron por triplicado para un total de 15 determinaciones. Se construyó la curva de calibración correspondiente a los resultados de los 15 puntos experimentales. Se aplicó el mismo procesamiento estadístico descrito para la linealidad del sistema, así como los mismos criterios de aceptación.
Exactitud: Se analizaron por triplicado placebos cargados con cantidades equivalentes al 80, 100 y 120 % con respecto a la cantidad teórica declarada. Se construyó una curva de recuperación de % recuperado (Y) vs. % añadido (X). Los resultados fueron procesados estadísticamente de igual forma que la curva de calibración de la linealidad del sistema y del método. Además se calculó el % de recobro (R) a través de la fórmula:
![]()
donde:
A: % de mefenesina recuperados
B: % de mefenesina teórico
Se incluyó la determinación del recobrado medio (![]() ) y del CV total.
) y del CV total.
Criterios de aceptación:
![]() : 98-102 %
: 98-102 %
CV £ 2,0 %
Además, se realizó la prueba G de Cochran, para determinar si el factor concentración tiene alguna influencia en los resultados. El valor de Gexp se calculó mediante la siguiente expresión:
![]()
Gtab (a= 0,05; k= 3; n= 3)
donde:
Smax: desviación estándar máxima
S1,2,3: desviación del nivel 1, 2 y 3
k: grupos experimentales
n: determinaciones por grupo
Si Gexp < Gtab las varianzas de las 3 concentraciones son equivalentes, o lo que es igual, el factor concentración no influye en la variabilidad de los resultados.
Por último, se aplicó la prueba de la t de Student para demostrar que no existen diferencias significativas entre el valor medio de recobro obtenido y el 100 %.
Siendo: n-1 los grados de libertad y a= 0,05
![]()
donde:
![]() : valor medio del % de recobro.
: valor medio del % de recobro.
CV: coeficiente de variación de la desviación típica del % de recobro,
n: número de determinaciones.
Selectividad: Se analizó una muestra de referencia de concentración equivalente al 100 %, registrando el cromatograma y estableciendo el tiempo de retención (tr) del analito. Se procesaron por triplicado placebos del producto aplicando el procedimiento descrito anteriormente. En cada caso se registró el cromatograma correspondiente. Además se procesaron por triplicado placebos cargados con el 100 % de analito y se calculó el porcentaje de recobro promedio obtenido, el cual se comparó estadísticamente con el 100 %, aplicando prueba de la t de Student de una cola (a = 0,05).
Criterio: El método es capaz de dar respuesta con el analito sin interferencia de los componentes de la matriz.
Precisión:
- Repetibilidad
Repetibilidad de la respuesta analítica al 100 %: Se evaluó por sextuplicado la muestra homogénea preparada a partir de la forma terminada y se calculó el CV. Criterio de aceptación: CV £ 2,0 %
Repetibilidad del método: Con los resultados obtenidos para la exactitud en cada caso, se estimaron los CV para cada nivel de concentración, los cuales se compararon con el límite establecido.
Precisión intermedia: Participaron dos analistas, en dos días diferentes, en el mismo laboratorio. Se analizaron por triplicado en cada caso muestras equivalentes al 100 % obtenidas a partir de la forma terminada. Se calculó el CV total. Criterio de aceptación: CV < 3,0 %
Además se realizaron las siguientes pruebas:
- Prueba F de Snedecor: Se utilizó para determinar si existían diferencias significativas entre los resultados de los analistas que emplearon el mismo método y entre los días en que se realizaron los análisis, calculándose según la siguiente expresión.
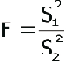
tal que F ³ 1
donde:
S1: Varianza del analista 1, o del día 1.
S2: Varianza del analista 2, o del día 2.
El valor de Fexp se comparó con Ftab para a = 0,05; F1= n-1 grados de libertad del numerador y F2= n - 1 grados de libertad del denominador.
Si Fexp < Ftab no existe diferencia significativa entre la precisión alcanzada por los analistas, en dos días de trabajo.
- Prueba de la t de Student: Se utilizó para comprobar si los valores obtenidos entre los analistas, que emplearon igual método, y los dos días en que realizaron los análisis eran homogéneos, para el nivel de significación a = 0,05 y los grados de libertad seleccionados F= (n1 + n2) - 2.
El valor de t se determinó mediante la siguiente ecuación:
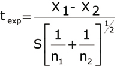
donde:
x1 y x2: valores de las medias de los análisis de las series 1 y 2, respectivamente.
S: varianza ponderada que se calcula como:
![]()
n1 y n2: número de determinaciones de las series 1 y 2, respectivamente.
El valor de texp se compara con el valor de la ttab para a= 0,05 y ƒ= n1 + n2 - 2.
Validación del método para el seguimiento de la estabilidad química de la mefenesina en las tabletas
Se evaluaron adicionalmente los parámetros requeridos por la categoría II:5
Selectividad: Se procesaron muestras de mefenesina materia prima sometidas a diferentes condiciones drásticas:
- Hidrólisis básica: 500 mg + 10 mL de solución de NaOH 1 N.
- Hidrólisis ácida: 500 mg + 10 mL de solución de HCl 1 N.
- Oxidación: 500 mg + 10 mL de H2O2 30 %.
- Humedad relativa (HR) de 85 %.
Además se prepararon placebos según el procedimiento establecido,3 los cuales fueron sometidos a iguales condiciones degradativas. De cada muestra se tomaron alícuotas de 0,1 mL, se trasvasaron a un matraz de 50 mL y se completó con fase móvil. Seguidamente se analizaron 20 µL, previamente filtrados. El tiempo máximo de exposición de las muestras fue de 2 h y 30 min.
Para cada muestra, se registró el cromatograma y se comparó con el de la solución de mefenesina (solución de referencia) preparada al 100 % tanto cualitativa como cuantitativamente.
Criterio: El método es capaz de dar respuesta con el analito selectivamente, en presencia de posibles productos de degradación sin interferencia.
Sensibilidad: La evaluación de este parámetro se realizó mediante la estimación del límite de cuantificación (Lc) y el límite de detección (Ld). Para ello se trabajaron con diferentes concentraciones; 10, 20 y 30 % equivalentes a 10, 20, 30 µg/mL respectivamente.
Se aplicaron los siguientes pasos:6
1. Se determinó la pendiente de la curva de calibración obtenida para la validación del método (AUC vs. concentración en %) y se obtuvo el valor de "b".
2. Se construyó otra curva de calibración analizando 3 puntos con 3 réplicas cada uno de concentraciones menores del analito 10, 20 y 30 µg/mL respectivamente) y se determinó la ecuación de esta nueva recta de calibración aplicando regresión lineal por el software Statistica for Windows (versión 6.01). Se extrapoló la respuesta a concentración cero y se obtuvo un estimado de la respuesta blanco "Y bl".
3. Se determinó la desviación estándar (S) correspondiente a cada concentración evaluada en el paso anterior. Se construyó otra curva de calibración de desviación estándar (S) vs. concentración (µg/mL). Al igual que en el paso anterior se extrapoló la S a concentración cero y se obtuvo el estimado "Slb" correspondiente a la S del blanco.
Se calculó Lc y Ld a través de las siguientes ecuaciones:
![]()
![]()
donde:
Ld: límite de detección
Lc: límite de cuantificación
b: pendiente de la recta de regresión lineal
Ybl: estimado del blanco
Slb: estimado de la desviación estándar del blanco
n: número de medidas individuales
Comprobación experimental del límite de cuantificación: Se analizaron por triplicado placebos cargados con concentración equivalentes a 0,58 µg. Se determinó Media y DE.
RESULTADOS
En la tabla 1 se resumen los principales resultados obtenidos en la validación del método, así como los criterios de aceptación, los cuales se cumplieron satisfactoriamente en cada caso.
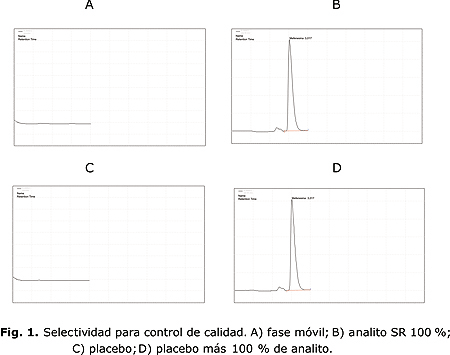
En la figura 1 se muestran los cromatogramas de la fase móvil (para control de calidad de la materia prima), del analito (SR), del placebo procesado por el método en estudio y del placebo en presencia del principio activo. El conjunto de estos resultados permitió afirmar que el método es selectivo para control de calidad.
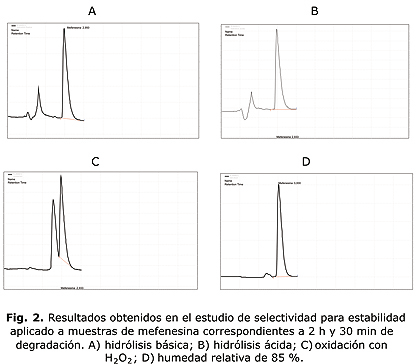
En la figura 2 se muestran los cromatogramas resultantes de la evaluación de la selectividad para estabilidad. Los resultados demostraron la adecuada selectividad del método frente a los componentes de la matriz, así como frente a los posibles productos de degradación.
Los valores utilizados en la estimación de los Ld y Lc se resumen en la tabla 2, así como los resultados obtenidos para estos parámetros. Una vez estimado teóricamente el Lc, se comprobó experimentalmente y se obtuvieron resultados satisfactorios. La recuperación promedio fue de 99,070 0,741 %. El método fue suficientemente sensible para el propósito con que fue desarrollado, pues es capaz de cuantificar una cantidad equivalente al 98,5 % de degradación.

DISCUSIÓN
El método fue lineal, por lo que el efecto de la matriz no fue significativo desde el punto de vista estadístico, resultados que están en perfecta correspondencia con los obtenidos en la selectividad para control de calidad.
En la exactitud al realizar el procesamiento estadístico, se comprobó el cumplimiento de todos los criterios exigidos según la regresión lineal aplicada. El coeficiente de recobrado medio no excedió el límite de 98-102 %. Además el CV total quedó comprendido en el rango de aceptación acotado por un límite superior igual al 2 %.
Adicionalmente se evaluó la influencia de la concentración de analito en la varianza (S) de los resultados a través de la prueba de G de Cochran. Como la Gexp < Gtab, las varianzas de los tres niveles de concentración evaluados, fueron equivalentes. Es decir, no influyó el factor concentración en la exactitud del método. Por su parte, la prueba de la t de Student corroboró la exactitud, ya que la t obtenida fue inferior a la tabulada, por lo que no existieron diferencias estadísticamente significativas entre el recobrado medio y el 100 %. De modo que se pueda afirmar que la técnica fue exacta y no se afectó por errores sistemáticos de forma significativa, por lo que permite obtener valores experimentales muy próximos al valor verdadero.
Para la repetibilidad de la respuesta analítica al 100 % se cumplió satisfactoriamente el criterio de aceptación establecido, por lo que la respuesta es repetible en condiciones similares de operación.
La repetibilidad del método se estimó mediante la determinación de los CV para 3 niveles de concentración: bajo, medio y alto. Se reportaron CV bajos, inferiores al 1,5 % establecido como límite, por lo que el método cumple con los criterios de aceptación establecidos y es suficientemente repetible para control de calidad de las tabletas.
Para la precisión intermedia también los valores de CV fueron inferiores al criterio de aceptación (CV £ 2,0 %), de modo que los errores aleatorios no repercutieron apreciablemente. EL análisis se complementó con las pruebas de Fischer y de la t de Student.
En ambos análisis la Fexp fue menor que la Ftab, por lo que no existieron diferencias significativas entre las precisiones de los analistas, independientemente del día en que se efectúo el ensayo. Los valores de las texp resultaron menores que las ttab en cada caso, por lo que no existieron diferencias significativas entre los valores medios obtenidos por los analistas. El conjunto de estos resultados permitió asegurar que estos fueron homogéneos, lo que ratificó la precisión del método en estudio para producto terminado.
Como se observa, el tratamiento aplicado y el procedimiento cromatográfico, garantizan la adecuada selectividad del método frente a la fase móvil empleada y a los componentes de la matriz para el caso de la formulación de tabletas, por lo que el método se considera selectivo y puede aplicarse al control de calidad de la mefenesina en las tabletas de producción nacional.
Como el objetivo fundamental de este método, es realizar el seguimiento de la estabilidad química de la mefenesina en las tabletas, se impone demostrar en la etapa de validación, que es un método suficientemente selectivo frente a los posibles productos de degradación, tanto del principio activo, como de los demás componentes de la formulación.
Por esta razón, fue necesario someter a condiciones degradativas al principio activo y a muestras placebo. Los placebos degradados no mostraron ninguna señal adicional, por lo que en todas las condiciones evaluadas, los cromatogramas fueron similares al presentado con anterioridad.
Para las muestras de mefenesina materia prima degradadas, los resultados mostraron adecuada separación del analito en todas las condiciones ensayadas, excepto para el medio oxidante (fig. 2, C). En este caso, solo se obtuvo una señal adicional atribuida al producto de degradación, por comparación del tr de la muestra degradada con la muestra de referencia de dicha sustancia en correspondencia con la disminución en la altura del pico del analito.
REFERENCIAS BIBLIOGRÁFICAS
1. Colectivo de autores. Principios de Química Farmacéutica. Parte I. La Habana, 1994. p. 261-71. (Edición Rrevolucionaria).
2. VIDAL. L'information de reference sur les produits de santé.iche descriptive abrégée du médicament : decontractyl 500 mg cp enr. Disponible en: http://www.vidal.fr/Medicament/decontractyl-4817.htm
3. Minh H. Estudios preliminares de reformulación de tabletas de mefenesina 500 mg. Tesis en opción al título de Licenciado en Ciencias Farmacéuticas. Instituto de Farmacia y Alimentos. Universidad de La Habana. La Habana, 2007.
4. Regulación No. 41-2006. CECMED. Validación de métodos analíticos.
5. USP 28- NF 23. United States Pharmacopoeia XXVIII and Formulary National 23st. United Stated Pharmacopeial Convention. Versión electrónica. 2005.
Recibido: 12 de enero de 2009.
Aprobado: 17 de febrero de 2009.
Dra. C. Yania Suárez Pérez. Instituto de Farmacia y Alimentos. Universidad de La Habana. Ave 23 No. 21 425 e/ 214 y 222, La Coronela, municipio La Lisa, La Habana, Cuba.


{kind=link}