










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Farmacia
versión On-line ISSN 1561-2988
Rev Cubana Farm v.43 n.4 Ciudad de la Habana dic. 2009
ARTÍCULOS ORIGINALES
Diseño y validación de un método espectrofotométrico para el control de calidad del piroxicam jalea 0,5 %
Design and validation of a spectrophotometry method used in quality control of a 0,5 % Piroxican jelly
Yania Suárez PérezI; Oscar García PulpeiroII; Thuy Dao ThanhIII; Munkhzul MishigIV
IDoctor en Ciencias Farmacéuticas. Licenciada en Ciencias Farmacéuticas. Máster en Tecnología y Control de Medicamentos. Profesor Auxiliar. Instituto de Farmacia y Alimentos. Universidad de La Habana. La Habana, Cuba.
IILicenciado en Ciencias Farmacéuticas. Máster en Tecnología y Control de Medicamentos. Empresa Laboratorio "Roberto Escudero Díaz". La Habana, Cuba.
IIILicenciado en Ciencias Farmacéuticas. República Popular de Mongolia.
IVLicenciado en Ciencias Farmacéuticas. República Socialista de Viet Nam.
RESUMEN
La jalea de piroxicam al 0,5 % es un nuevo producto en fase de desarrollo de la Empresa Laboratorio "Roberto Escudero Díaz". En el presente trabajo se diseñó un método por espectrofotometría UV, para lo cual se empleó ácido clorhídrico etanólico como disolvente y 328 nm como longitud de onda de máxima absorción. Se estableció un procedimiento simple y rápido para procesar la muestra previa al análisis. Posteriormente se aplicó la validación de este según exigencias regulatorias actuales. El método resultó suficientemente específico, lineal, preciso y exacto en el rango de 3,125 a 9,375 µg/mL. Se aplicó a la cuantificación del principio activo en 3 lotes de la formulación desarrollada, con resultados satisfactorios sin diferencias estadísticamente significativas según el procesamiento aplicado.
Palabras clave: Piroxicam, jalea, espectrofotometría, validación.
ABSTRACT
The 0.5 % jelly Piroxican is a new product in developing phase from "Roberto Escudero Díaz" Laboratory Enterprise. In present paper a UV spectrophotometry method was designed using ethanol chloride acid as solvent and 328 nm as maximal absorption wave length. A simple and fast procedure was established to processing the sample previous to analysis. Subsequently, we applied the validation of it according current regulatory demands. Method was enough specific, linear, precise and exact at the 3.125 to 9.375 µg/mL. Active principle quantification was applied in three batches of developed formula achieving satisfactory results without statistically significant differences according the applied procedure.
Key words: Piroxican, jelly, spectrophotometry, validation.
INTRODUCCIÓN
La cuantificación de piroxicam en cualquier matriz por métodos oficiales se realiza por cromatografía líquida de alta resolución (CLAR) y detector ultravioleta (UV) a 248 nm.1,2
La aplicación de la espectrofotometría UV se limita al ensayo de disolución de las cápsulas de piroxicam.1,2 La solución de la muestra previamente filtrada se analiza por comparación contra patrón a 333 nm.
Teniendo en cuenta las posibilidades desde el punto de vista analítico que presenta la estructura de este compuesto para su cuantificación por métodos espectrofotométricos, la facilidad de aplicación de estas técnicas en cualquier laboratorio, así como la importancia de desarrollar un método confiable pero a la vez, rápido, sencillo y de bajo costo para asumir el control de rutina de la nueva formulación una vez introducida, se valoró como una alternativa ventajosa para el control de calidad del piroxicam en la jalea al 0,5 %.
MÉTODOS
Desarrollo de un método espectrofotométrico para control de calidad del piroxicam en la jalea al 0,5 %
Se diseñó un método por espectrofotometría UV directo.
- Determinación de la longitud de onda de máxima absorción del piroxicam en solución
En la etapa de desarrollo del método se determinó la longitud de onda (l) de máxima absorción del piroxicam materia prima en solución. Se preparó una solución de concentración 6,25 µg/mL, con el empleo de HCl etanólico 0,01 N como disolvente, a la cual se le determinó el espectro UV en el rango de 220 a 430 nm.
- Procesamiento de la muestra para el análisis
Teniendo en cuenta que el método se diseña para el control de calidad del piroxicam en la jalea al 0,5 %, fue necesario proponer una metodología analítica que permitiera obtener una solución del principio activo de concentración adecuada a partir de la forma terminada.
A continuación se describe el método propuesto:
1. Pesar 0,125 g de jalea de piroxicam 0,5 % en un vaso de precipitado de 50 mL.
2. Añadir 10 mL de ácido clorhídrico etanólico 0,01 N y agitar vigorosamente.
3. Trasvasar cuantitativamente a matraz de 100 mL.
4. Completar a volumen con ácido clorhídrico etanólico 0,01 N y agitar.
5. Leer la absorbancia de la muestra a la longitud de onda de máxima absorción empleando el disolvente como ensayo de corrección.
Validación del método analítico para control de calidad
Se realizó la validación del método espectrofotométrico según los parámetros mínimos establecidos para la categoría I.2 Además se tuvieron en cuenta las exigencias regulatorias nacionales.3
Especificidad: Se evaluaron por triplicado placebos del producto aplicando el método propuesto. Se registró el espectro UV en el rango de 220-370 nm y se realizaron lecturas de estas muestras por triplicado a l = 328 nm.
Criterio de aceptación: No se debe registrar respuesta en el rango de interés analítico para el piroxicam.
Linealidad del sistema: Se realizó mediante el análisis de 5 concentraciones de piroxicam materia prima por triplicado, en un rango de 50-150 % de la cantidad teórica declarada como 100 % (6,25 µg/mL), equivalentes a 3,125; 4,687; 6,250; 7,8125 y 9,375 µg/mL respectivamente.
Se construyó una curva de calibración de absorbancia vs. concentración teórica (%). Los resultados se procesaron estadísticamente a través del paquete STATISTICA for Windows versión 6.01 (opción regresión lineal múltiple) y se determinó: coeficiente de correlación lineal (r), coeficiente de determinación (r2), intercepto (a) y pendiente (b), para el 95 % de confianza
Criterios de aceptación:
- Ecuación de la recta: y= bx + y
- ³ 0,99
- ³ 0,98
- Prueba de proporcionalidad del método analítico o hipótesis nula de la ordenada en el origen a= 0.
- Se empleó la prueba estadística de la t de Student para n-2 grados de libertad, siendo n el número total de valores donde: texp < ttab.
- Prueba de la hipótesis nula de la pendiente: b= 0. Se determinó a partir de una prueba ANOVA de la regresión, teniendo en cuenta la probabilidad asociada al valor de la pendiente, es decir, si la p<< 0,05, el valor de "b" difiere significativamente de cero.
- Se calcularon los factores de respuesta (ƒ), el valor medio, la DE y el coeficiente de variación (CV) que debe ser menor que 5 %.4
Linealidad del método
Se evaluaron placebos del producto cargados con el 50, 75, 100, 125 y 150 % del principio activo respectivamente. En cada nivel de concentración se analizaron 3 muestras. Se construyó la curva de calibración correspondiente a los resultados de los 15 puntos experimentales. Se aplicó el mismo procesamiento estadístico descrito para la linealidad del sistema, así como los mismos criterios de aceptación.
Exactitud: Se analizaron por triplicado placebos cargados con cantidades equivalentes al 50, 100 y 150 % respecto a la cantidad teórica declarada. Se construyó una curva de recuperación de % recuperado (Y) vs. % añadido (X). Los resultados fueron procesados estadísticamente de igual forma que la curva de calibración de la linealidad del sistema y del método. Además se calculó el % de recobro (R), el recobrado medio (![]() ) y el CV total.
) y el CV total.
Criterios de aceptación: ![]() : 96-104 % (para productos semisólidos) y CV £ 3,0 %
: 96-104 % (para productos semisólidos) y CV £ 3,0 %
Además, se realizó la prueba G de Cochran, para determinar si el factor concentración tiene alguna influencia en los resultados. Si Gexp < Gtab las varianzas de las 3 concentraciones evaluadas son equivalentes, o lo que es igual, el factor concentración no influye en la variabilidad de los resultados.4
Por último se aplicó la prueba de la t de Student para demostrar que no existen diferencias significativas entre el valor medio de recobro obtenido y el 100 %, siendo: n-1 los grados de libertad y a = 0,05.4,5
Precisión
- Repetibilidad.
Para la repetibilidad se evaluaron por triplicado placebos cargados con la concentración equivalente al 100 % y un valor bajo y otro alto comprendido dentro del rango de la linealidad del método, 50 y 150 % respectivamente. Se calculó el CV en estos 3 niveles de concentración y se comparó con el criterio establecido. Las determinaciones las realizó el mismo analista en las mismas condiciones de trabajo.
Criterio de aceptación: CV £ 1,5 %.
- Precisión intermedia
Participaron 2 analistas, en 2 días diferentes, en el mismo laboratorio. Se analizaron por triplicado en cada caso muestras equivalentes al 100 %. Se calculó el CV total.
Criterio de aceptación: CV < 3,0 %
Además se realizaron las siguientes pruebas:
- Prueba F de Snedecor: Se utilizó para determinar si existían diferencias significativas entre los resultados de los analistas que emplearon el mismo método y entre los días en que se realizaron los análisis. El valor de Fexp se comparó con Ftab para a = 0,05; F1= n - 1 grados de libertad del numerador y F2= n -1 grados de libertad del denominador. Si Fexp < Ftab no existe diferencia significativa entre la precisión alcanzada por los analistas, en 2 días de trabajo.4,6
- Prueba de la t de Student: Se utilizó para comprobar si los valores obtenidos entre los analistas, que emplearon igual método, y los 2 días en que realizaron los análisis eran homogéneos, para el nivel de significación a = 0,05 y los grados de libertad seleccionados F= (n1 + n2) - 2. Si valor de texp < ttab, no existen diferencias significativas.4,6
Rango
Se estableció el intervalo en que se cumplen los criterios de linealidad, exactitud y precisión del método.
Control de calidad de los lotes elaborados a escala de laboratorio y de la formulación de referencia
A cada lote de producto terminado se le realizó el control químico con el empleo del método espectrofotométrico diseñado y validado para el control de rutina de los lotes de jalea de piroxicam al 0,5 %.
Los resultados obtenidos se compararon estadísticamente con el empleo del análisis de varianza de clasificación simple a través del programa ANOVA-1 (López R, 1994). La existencia de diferencias significativas se determinó para el 95 % de confianza.
RESULTADOS
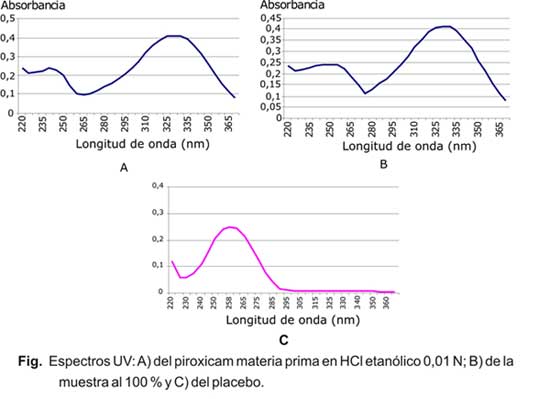
El espectro UV del piroxicam materia prima en HCl etanólico 0,01 N, se muestra en la figura (A). Se observaron 2 máximos: 240 y 328 nm. Se seleccionó para la cuantificación la lmáxima = 328 nm.
Una vez seleccionada la l de máxima absorción y establecida una concentración de fármaco cuya absorbancia fue adecuada (concentración de 6,25 µg/mL con valores de absorbancia alrededor de 0,45), se procedió a establecer una metodología analítica muy sencilla que permitiera obtener una solución transparente a partir de la forma terminada. En la figura (B) se presenta el espectro UV de una muestra al 100%.
El primer parámetro analizado durante la etapa de validación fue la especificidad. En la figura (C) se muestra el espectro UV del placebo tratado por el método propuesto. Como se observa la absorción UV del placebo ocurre a l menores de 290 nm, atribuidas al metil y propilparabeno presentes en la formulación. El método resultó específico ya que los valores de absorbancia obtenidos en el rango de interés analítico para el piroxicam, no constituyen interferencias significativas (0,0056 ± 0,0005), ya que equivalen a menos del 1 %.
Los resultados de la linealidad del sistema y del método se muestran en la tabla 1 y los correspondientes a repetibilidad y exactitud se resumen en la tabla 2.


La linealidad en ambos casos fue satisfactoria según el cumplimiento de los parámetros estadísticos establecidos (tabla 3).
Al realizar el procesamiento estadístico de los resultados de la exactitud, se comprobó el cumplimiento de todos los criterios de aceptación exigidos (tabla 3). La técnica fue exacta y no se afectó por errores sistemáticos de forma significativa.
Para la repetibilidad se estimó el CV para 3 niveles de concentración: bajo, medio y alto. Se reportaron CV bajos, inferiores al 1,5 % establecido como límite (tablas 2 y 3).
La precisión intermedia también fue evaluada y los resultados fueron satisfactorios. Se obtuvo un CV acorde con el criterio de aceptación (CV £ 3,0 %). Además se cumplieron satisfactoriamente las restantes pruebas aplicadas, de modo que los errores aleatorios no repercutieron en gran medida sobre el método desarrollado.
Todos los lotes mostraron correcta dosificación de piroxicam, según criterios de Farmacopea1 (tabla 4). La comparación estadística entre formulaciones por análisis de varianza se realizó por ANOVA-1. Los resultados se reflejan en la tabla 4, los cuales demostraron que no existen diferencias ni entre lotes ni entre réplicas, por lo que se garantizó reproducibilidad en la dosificación, que además cumple con el rango establecido de 90-110 %.
DISCUSIÓN
Se desarrolló un método espectrofotométrico directo para el control de calidad del piroxicam en la jalea al 0,5 %, teniendo en cuenta la presencia de grupos cromóforos en la estructura del compuesto. La espectrofotometría UV tiene como ventajas la rapidez y simplicidad de la metodología analítica a seguir. Con vistas al posterior registro de este producto por la entidad reguladora nacional, se recomienda el uso de este método espectrofotométrico para la liberación de los lotes de forma más rápida.
Se seleccionó para la cuantificación la lmáxima = 328 nm, ya que en esta zona y en el medio ligeramente ácido utilizado, no se deben presentar interferencias de los parabenos. La absorción de los parabenos en medio ácido se desplaza hacia menores longitudes de onda, por efecto hipsocrómico, es decir, se presenta por debajo de los 300 nm. Este fenómeno sería una complicación desde el punto de vista analítico si se trabajara en medio alcalino, donde ocurre efecto batocrómico de la banda de absorción de los parabenos.
El empleo del HCl etanólico 0,01 N como disolvente, se justifica teniendo en cuenta la pobre solubilidad del principio activo en medio acuoso y en etanol. A su vez se reporta como disolvente del piroxicam en Farmacopea, el HCl metánolico de igual concentración a la seleccionada, para la identificación por cromatografía en capa delgada (CCD), para la preparación de las muestras y patrones en el método de cuantificación por CLAR en la jalea y en el ensayo de disolución de las cápsulas de piroxicam.1 Se sustituyó el metanol, disolvente de mayor costo y toxicidad por el alcohol etílico de producción nacional. De esta forma se propone una metodología menos agresiva para el hombre y el medio ambiente, y al mismo tiempo, más rentable desde el punto de vista económico.
El conjunto de resultados obtenidos en los ensayos de linealidad, permitió afirmar que tanto el sistema como el método fueron lineales en el rango estudiado (3,125-9,375µ/mL), ya que se obtuvo una elevada proporcionalidad entre la respuesta obtenida y la concentración del analito, avalado por el cumplimiento de todos los criterios estadísticos que derivan del análisis de la regresión lineal.
El recobrado medio no excedió el límite de 96-104% propuesto para preparaciones semisólidas. Además el CV total quedó comprendido en el rango de aceptación acotado por un límite superior igual al 3%. Adicionalmente se evaluó la influencia de la concentración de analito en la varianza (S) de los resultados a través de la prueba de G de Cochran (tabla 3). Como la Gexp < Gtab, las varianzas de los 3 niveles de concentración evaluados fueron equivalentes. Es decir, no influyó el factor concentración en la exactitud del método. Por su parte, la prueba de la t de Student corroboró la exactitud, ya que la t obtenida fue inferior a la tabulada, por lo que no existieron diferencias estadísticamente significativas entre el recobrado medio y el 100 %.
En el estudio de la precisión, el análisis se complementó con las pruebas de Fischer y de la t de Student. En ambos análisis la Fexp < Ftab por lo que no existieron diferencias significativas entre las precisiones de los analistas, independientemente del día en que se efectuó el ensayo. Los valores de las texp resultaron menores que las ttab en cada caso, por lo que no existieron diferencias significativas entre los valores medios obtenidos por los analistas. El conjunto de estos resultados permitió asegurar que los resultados fueron homogéneos, lo que ratifica la precisión del método en estudio.
El cumplimiento de las exigencias establecidas para la validación de técnicas analíticas, garantiza que el procedimiento desarrollado fue suficientemente específico, lineal, preciso y exacto para la cuantificación del piroxicam en la jalea al 0,5 %, por lo que se considera un método válido para realizar el control de rutina de este nuevo medicamento, en el rango estudiado.
REFERENCIAS BIBLIOGRÁFICAS
1. British Pharmacopoeia. 2004. London: The Stationery Office. CD ROM.
2. USP 30, 2007. United States Pharmacopoeia 30 and National Formulary 25. The official compendia of standard. Versión Electrónica.
3. CECMED. Regulación 41-2007. Validación de Métodos analíticos. 2007.
4. Pérez N. Curso Validación de técnicas de análisis químico. Enfoque práctico. Centro de Ingeniería e Investigaciones Químicas. La Habana, 14-18 noviembre, 2003.
5. ICH Q2B. Harmonised Tripartite Guideline. Validation of analytical procedures: Methodology. International Conference on Harmonisation of Technical Requeriments for registration of pharmaceuticals for human use. Nov., 1996.
6. Gil AE. Protocolo de Validación de Métodos de análisis: Materias prima y producto terminado. Facultad de Farmacia. Universidad Complutense de Madrid, España. 1995.
Recibido: 16 de julio de 2009.
Aprobado: 20 de agosto de 2009.
Lic. Yania Suárez Pérez. Instituto de Farmacia y Alimentos. Universidad de La Habana. Ave 23 No. 21 425 e/ 214 y 222, La Coronela, municipio La Lisa, La Habana, Cuba. Correo electrónico: yania_as@yahoo.es


{kind=link}