










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Farmacia
versión On-line ISSN 1561-2988
Rev Cubana Farm v.43 n.4 Ciudad de la Habana dic. 2009
ARTÍCULOS ORIGINALES
Método analítico para la cuantificación y ensayo de disolución de risperidona tabletas 3 mg
Analytical method for quantification and dissolution essay of 3 mg tablets of Risperidone
Caridad Margarita García PeñaI; Lissette Martínez MirandaII;Antonio Iraizoz BarriosIII; Vivian Martínez EspinosaIV; Matilde Torres GarcíaIV; Gissel María León GuerreroV
ILicenciada en Ciencias Farmacéuticas. Máster en Tecnología y Control de Medicamentos. Investigadora Agregada. Centro de Investigación y Desarrollo de Medicamentos (CIDEM). La Habana, Cuba.
IILicenciada en Bioquímica. Investigadora Auxiliar. CIDEM. La Habana, Cuba.
IIILicenciado en Ciencias Farmacéuticas. Máster en Tecnología y Control de Medicamentos. Investigador Agregado. CIDEM. La Habana, Cuba.
IVTécnico en Tecnología Farmacéutica. CIDEM. La Habana, Cuba.
VLicenciada en Química. CIDEM. La Habana, Cuba.
RESUMEN
Se desarrolló un método analítico por cromatografía líquida de alta resolución para la cuantificación y el ensayo de disolución de las tabletas de risperidona 3 mg. El método se basó en la separación del principio activo a través una columna cromatográfica Lichrosorb RP-18 (5 µm) (250 x 4 mm), con detección UV a 278 nm, para lo cual se empleó una fase móvil compuesta por acetonitrilo:buffer fosfato de potasio 0,05 M, de proporción 45:55. El método para la cuantificación del principio activo fue validado a través de la linealidad del sistema, especificad, exactitud y precisión. Mientras que en la validación del ensayo de disolución se evaluó la linealidad, precisión, especificidad e influencia del filtrado. Ambos métodos fueron sencillos, rápidos y económicos; además de específicos, lineales, precisos y exactos en el rango de concentraciones estudiadas. El método analítico alternativo desarrollado para la cuantificación y disolución de las tabletas de risperidona 3 mg, se comparó estadísticamente con el método propuesto en la Farmacopea de los Estados Unidos 30, y se demostró que no existían diferencias significativas entre los resultados obtenidos por cada método.
Palabras clave: Risperidona, disolución, cuantificación, validación, cromatografía líquida de alta resolución.
ABSTRACT
A high-performance liquid chromatography analytical method was developed for quantification and in dissolution assay of 3 mg Risperidone tablets. Method was based in active principle separation through a Lichrosorb RP-18 (250 x 4 mm) chromatographic column with UV detection to 278 nm using a mobile phase composed of 0.05 potassium phosphate buffer:acetonitrile, of 45:55 ratio. Method for active principle quantification was validated through linearity, specificity, precision and accuracy of system. Whereas in dissolution assay validation the linearity, precision, specificity and filtrate influence was assessed. Both methods were simple, fast and economic as well as specific, linear, precise and exact within the study concentrations rank. The alternative analytical method developed for 3 mg Risperidona tablets quantification and dissolution was statistically compared to method proposed by USA Pharmacopeia demonstrating that there were not significant differences among the results achieved by each method.
Key words: Risperidone, dissolution, quantification, validation, high-performance liquid chromatography.
INTRODUCCIÓN
La risperidona (C23H27FN4O2 , PM= 410,49), cuya denominación química es 4H-pirida[1,2-a]pirimidina-4-ona,3-[2-[4-(6-fluoro-1,2bencioxazol-3-y1)-1-piperidinil]etil]-6,7,8,9 -tetrahidro2metil, es un antipsicótico benzisoxazólico, reportado como antagonista de receptores de dopamina D2 y serotonina (5-HT2), adrenérgicos (a1 y a2) e histamina (H1). Se indica en el tratamiento de la psicosis crónica y aguda y de la manía.1-3
En la Farmacopea de los Estados Unidos (USP 30, 2007), se reporta un método analítico por cromatografía líquida de alta resolución (CLAR) para la cuantificación del principio activo, con ácido trifluoroacético, teniendo en cuenta que presentaban problemas con la disponibilidad de este en el Centro de Investigación y Desarrollo de Medicamentos (CIDEM), se desarrolló un método alternativo.4
El desarrollo de técnicas o métodos analíticos novedosos o la adecuación de algunos ya reportados es un problema cotidiano. Siempre que esto suceda se exige como parte integral del estudio, la validación del método en cuestión. La validación proporciona un alto grado de confianza y seguridad del proceso productivo o del método analítico, así como también en la calidad de los resultados.
Los parámetros analíticos que pueden ser considerados en la validación de un método analítico, según se expresa en USP 30, son exactitud, precisión, especificidad, límite de detección, límite de cuantificación, linealidad, rango, tolerancia y robustez.4
La CLAR le brinda la posibilidad al analista de emplear esta herramienta para solucionar los inconvenientes de los métodos espectrofotométricos en los estudios de estabilidad, pues además de presentar una alta sensibilidad y exactitud, es en esencia un método separativo, lo que permite medir con gran selectividad el compuesto deseado, siempre y cuando se encuentre un sistema cromatográfico que asegure una adecuada separación.5,6
El ensayo de disolución constituye uno de los estudios más importantes en el desarrollo de una forma farmacéutica sólida oral. Permite evaluar los procesos de fabricación, la calidad intra e interlotes, además de predecir en algunos casos la bioequivalencia y biodisponibilidad de productos sólidos orales y la estabilidad del preparado farmacéutico. 7
Como en todo método analítico, es indispensable efectuar la validación de este, para facilitar la utilización rutinaria con la fiabilidad adecuada. La USP 30, 2007, plantea para el ensayo de disolución (categoría III) el estudio de la precisión, mientras que los restantes parámetros se realizarán en dependencia de la naturaleza específica del ensayo. Sin embargo, en otros reportes o fuentes se incluyen aspectos tan importantes como: la especificidad con respecto al placebo, la linealidad y precisión y la influencia de la filtración.4-8
El objetivo de este trabajo fue el desarrollo y validación de un método analítico cromatográfico para la cuantificación del principio activo en la tableta y para la realización del ensayo de disolución de risperidona tabletas 3 mg. Además, se propuso demostrar si existen diferencias significativas entre el método desarrollado y el reportado en la USP 30, para la cuantificación y ensayo de disolución del producto terminado.
MÉTODOS
La sustancia de referencia química de risperidona fue suministrada por el grupo de sustancias de referencia del CIDEM, el cual fue analizado por el método cromatográfico establecido para realizar el control de la calidad de la materia prima, con una pureza de 99,8 %. El producto terminado en forma de tabletas, fue elaborado en la UCTB Tecnologías básicas, perteneciente al CIDEM, identificado como el lote 6001, fabricado en noviembre de 2006, el cual cumplió con las especificaciones de calidad establecidas para el control de la calidad de las tabletas.
Todos los reactivos utilizados fueron de grado HPLC, procedentes de la Riedel-de Haen.
Desarrollo del método analítico para la cuantificación del principio activo
En el desarrollo del método analítico por cromatografía, se utilizó un cromatógrafo (KNAUER) con detector UV/VIS (KNAUER) ajustado a 278 nm, un dosificador (loop) de 20 µL e integrador (SHIMADZU CR 6 A). La separación se realizó isocráticamente sobre una columna Lichrosorb (RP-18) 250 x 4 mm (5 µm). La fase móvil utilizada consistió en una mezcla desgasificada de acetonitrilo:buffer fosfato de potasio 0,05 M (45: 55) con una velocidad de flujo de 1,5 mL/min.
La solución de referencia se obtiene disolviendo risperidona sustancia de referencia, en ácido clorhídrico 0.1 N, hasta obtener una concentración de 60 µg/mL aproximadamente.
En la preparación de la solución de ensayo para la valoración se disuelve una cantidad exacta de polvo de tabletas equivalentes a 3 mg de risperidona en ácido clorhídrico 0,1 N hasta una concentración de alrededor de 60 µg/L.
Validación del método analítico para la cuantificación del principio activo
La validación fue realizada según la categoría I (USP 30) y la Regulación 41-2007 del Centro para el Control Estatal de la Calidad de los Medicamentos (CECMED), para la validación de métodos de análisis; se evaluaron los parámetros que a continuación se describen:4,9
Linealidad
Para el análisis de la linealidad se realizó el modelo de 3 determinaciones para 5 concentraciones diferentes, que representan el 60, 80, 100,120, y 140 % de la cantidad teórica declarada, y se determinó la ecuación de la recta, el coeficiente de correlación, la prueba de significación estadística de significación de la pendiente Sb rel(%), los coeficiente de variación de los factores de respuesta y ensayo de proporcionalidad.
Exactitud
Para el análisis de la exactitud se realizó el modelo de 3 réplicas, para 3 concentraciones diferentes: 80, 100, 120 %; se determinó el % de recuperación, la desviación estándar y el coeficiente de variación. Se determinó además la prueba de G de Cochran con vistas a comprobar si la variación de la concentración produce diferencias significativas en los resultados y la prueba de la t de Student para determinar diferencias significativas entre la recuperación media y el 100 %.
Precisión
Se evaluó la precisión del método mediante el estudio de repetibilidad y precisión intermedia. La repetibilidad se estudió sobre la base de 6 determinaciones y se determinaron los valores medios, la desviación estándar y el coeficiente de variación. El estudio de precisión intermedia se realizó por 2 analistas, en 3 días diferentes, a 3 niveles de concentración (que representan el 80, 100, 120 % de la concentración teórica), mediante la aplicación de la prueba de Fisher y de la t de Student para determinar si existían diferencias significativas entre los resultados al variar las condiciones de análisis.
Especificidad
Para el estudio de especificidad se analizaron: la sustancia de referencia de risperidona, el placebo, las muestras de producto terminado y muestras sometidas a condiciones drásticas como: hidrólisis ácida HCl 5 N, hidrólisis básica NaOH 5 N y oxidación H2O2, para determinar si existían interferencias de los excipientes de la formulación o de los productos de degradación en la determinación del principio activo.
Validación del método analítico para el ensayo de disolución10-14
Se emplearon las mismas condiciones cromatográficas que las de la cuantificación del principio activo en las tabletas de 3 mg, por CLAR, solo se modificó el dosificador empleado (loop) que en el ensayo de disolución fue de 200 µL.
En el ensayo de disolución se empleó un disolutor ERWEKA de 6 plazas, para lo cual se utilizaron las siguientes condiciones:
- Volumen: 500 mL.
- Aparato 2: paleta.
- Velocidad: 75 rev/min.
- Tiempo: 30 min.
- Medio de disolución: ácido clorhídrico 0,1 N.
Para el análisis se disolvió una cantidad exacta del estándar de referencia con ácido clorhídrico de modo que se obtuviera una solución con una concentración final de 6 µg/mL. Las muestras fueron preparadas de la siguiente manera: se añadió la tableta en cada una de las plazas del disolutor bajo las condiciones ya descritas, luego de transcurrido el tiempo de duración del ensayo, se extrajo una alícuota de 10 mL la cual fue filtrada y posteriormente se procedió a la lectura en el cromatógrafo.
Especificidad
Este estudio se realizó con el empleo de 6 unidades del placebo y según el procedimiento descrito para el ensayo de disolución, con el objetivo de determinar las posibles interferencias de los excipientes de la formulación en la determinación del principio activo. Debe cumplirse que no debe observarse interferencia de los excipientes de la formulación en la zona de elusión del principio activo.
Linealidad
Para el estudio de la linealidad de la respuesta del detector se preparó una curva de calibración con soluciones patrón de risperidona en un rango de concentraciones del 60 al 140 % de la cantidad teórica declarada. Debe lograrse un coeficiente de correlación (r) ³ 0,999; un coeficiente de variación de los factores de respuesta (CVf ) £ 5 %; y la prueba de significación del intercepto, mediante la prueba de la t de Student, la t calculada (t cal) debe ser menor que la t tabulada (t tab).
Precisión
La precisión del ensayo de disolución se estudió mediante 6 ensayos de disolución en los que se evaluaron las tabletas en 2 días diferentes. Debe cumplirse para un método lineal que los valores de t calculada de cada muestra fueran menores que el valor de t tabulada, para g.l.: 5; p < 0,05.
Influencia de la filtración
Se realizaron 6 ensayos de disolución y se compararon entre sí los resultados de las muestras filtradas por papel y las muestras centrifugadas a 3 000 rev/min durante 10 min. Debe cumplirse que los valores de t calculada para cada una de las muestras, al compararse estadísticamente los resultados obtenidos para cada uno de los medios filtrantes, deben ser menores que el valor de t tabulada.
Estabilidad del principio activo
La estabilidad del principio activo de risperidona, en forma de tabletas, en el medio de disolución se determinó con la realización de mediciones antes y después del tratamiento de las muestras a 37 ºC durante 1 h. Debe cumplirse que los valores del intercepto y la pendiente después del tratamiento deben encontrarse incluidos en los intervalos del intercepto y la pendiente de la curva de calibración inicial.
Comparación estadística
Con el objetivo de realizar una comparación estadística entre los resultados por la USP 30 y los obtenidos por el método analítico alternativo desarrollado, se aplicó el estadígrafo de la t de Student para determinar si existían diferencias significativas entre ambos métodos, para lo que se empleó el procesador estadístico Minitab 14.0.
RESULTADOS
Validación del método analítico para la cuantificación del principio activo
La figura muestra los resultados obtenidos en el estudio de especificidad del método. Como se observa en el cromatograma correspondiente a la muestra placebo (A) no se obtuvo ninguna señal en la zona de interés, al ser comparado con la señal obtenida para la solución estándar de referencia (B) y de la muestra de risperidona tabletas (C), lo cual indica que los excipientes o sustancias auxiliares de la solución no interfieren en la determinación del principio activo. En cuanto a las muestras sometidas a condiciones drásticas de: hidrólisis básica, hidrólisis ácida, oxidación (D, E, F), se detectó en el caso de la hidrólisis ácida y básica una pequeña disminución del pico del principio activo sin la aparición de picos secundarios que interfieran, al igual que en el caso de la oxidación con peróxido.
En el estudio de la linealidad del sistema se obtuvo la ecuación de la recta que se expresa según: y= -2,7674 + 0,58741 X. El coeficiente de correlación lineal fue de 0,99335. Al aplicar la prueba de linealidad, con la utilización del coeficiente de variación de los factores de respuesta, se obtuvo un CVf = 3,45 %.
El resultado del estudio de precisión intermedia del método desarrollado aparece reportado en la tabla 1. En el estudio de repetibilidad realizado, la media obtenida fue de 98,15 % y el coeficiente de variación fue de 0,79 %.
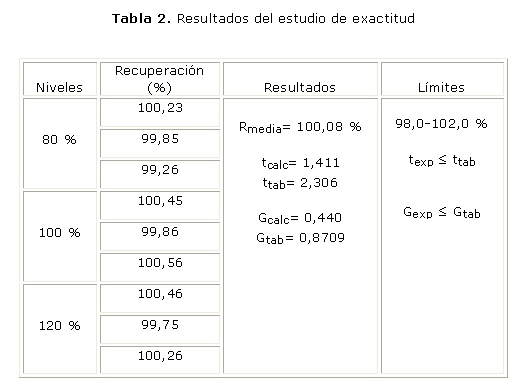
En la tabla 2, aparecen reportados los resultados del estudio de exactitud.
Validación del método analítico para el ensayo de disolución.
Los resultados que se obtienen en el estudio de especificidad, permiten afirmar que el método es específico en relación con los excipientes que componen la tableta porque no se observa interferencia de estos en la zona de elución del principio activo.
Al aplicar el método de los mínimos cuadrados, en el estudio de linealidad, a los resultados registrados, se obtuvo la ecuación de la recta que se expresa según: y= 0,0479 X + 0,1247. El coeficiente de correlación lineal fue de 0,99911. Al aplicar la prueba de linealidad, con la utilización del coeficiente de variación de los factores de respuesta, se obtuvo un CVf = 4,13 %, y al realizar la prueba de significación de la pendiente, el valor de t calculada (t cal= 2,01) resultó menor que la t tabulada (t tab= 2,14).
En la tabla 3 se muestran los resultados del estudio al realizar diferentes ensayos de disolución con el objetivo de analizar la disolución de las tabletas de risperidona, en 2 días diferentes. Se reportan los valores de las medias obtenidas en los ensayos de disolución realizados en los días diferentes, además los valores de la prueba de la t de Student calculados para cada muestra.

Los resultados de la influencia de la filtración aparecen reportados, en la tabla 4, para lo cual se emplearon 2 medios de filtración diferentes.

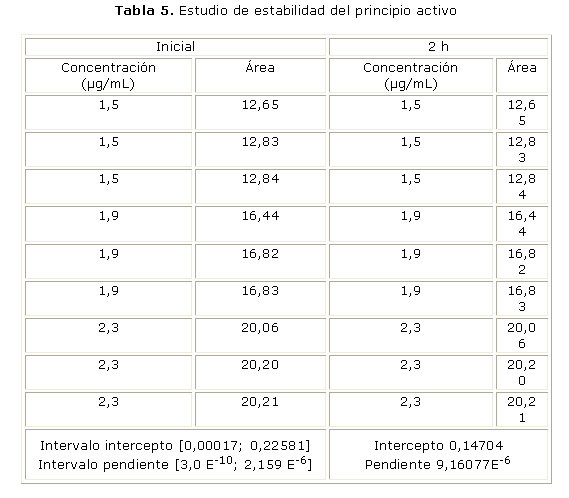
Los resultados del estudio de la estabilidad del principio activo en el medio de disolución, se presentan en la tabla 5.
Comparación estadística
Los resultados obtenidos al aplicar la prueba de la t de Student en la comparación estadística realizada a los métodos analíticos, para la valoración como para el ensayo de disolución, fueron 0,60 y 0,87 respectivamente, para una t tabulada de 2,33.
DISCUSIÓN
Validación del método analítico para la cuantificación del principio activo
Los resultados obtenidos en el estudio de especificidad del método (figura 2), demuestran la especificidad del método porque no existen interferencias de los excipientes y productos de degradación, al detectar separadamente el principio activo de sus excipientes y sin interferencias de los posibles productos de degradación. Estos resultados permiten asegurar la especificidad del método analítico alternativo desarrollado para el control de la calidad y estudios de estabilidad de las tabletas de risperidona 3 mg.
Los resultados del estudio de linealidad muestran coeficientes de regresión y de determinación superiores a los exigidos, 0,99 y 0,98 respectivamente, lo que demuestra con el valor del coeficiente de correlación obtenido, cercano a la unidad, la existencia de correlación con una probabilidad elevada, así como el grado de relación entre las variables concentración y respuesta detectada por el equipo empleado. También el coeficiente de variación de los factores de respuesta y la desviación estándar relativa de la pendiente fueron inferiores al normado como máximo para estos indicadores: 5 y 2 %, respectivamente, ambos son considerados estimadores puntuales que permiten caracterizar la variabilidad.4,9 El valor obtenido del CVf permite demostrar que existe variabilidad en la relación respuesta y concentración para cada nivel evaluado. El intervalo de confianza del intercepto incluye al cero, lo que permite excluir la significación del error del intercepto, y se demuestra con los resultados obtenidos la linealidad del método propuesto.
En el estudio de la repetibilidad realizado a una misma muestra, por el mismo analista, el mismo día, a través de 6 réplicas, se alcanzó un coeficiente de variación adecuado (0,79 %), lo que demuestra la buena precisión del método, y se observa una variabilidad de los resultados dentro de los límites establecidos para los métodos cromatográficos: CV £ 2,0 %.9
Los valores que se obtienen en el estudio de precisión intermedia, de las pruebas de Fischer y de la t de Student, demuestran que no existen diferencias significativas entre las precisiones alcanzadas por los analistas en diferentes días para una probabilidad de 0,05 %, ya que el valor de Fcalculada es menor que la Ftabulada, este resultado permite establecer que las precisiones son similares (tabla 1). Al realizar la prueba de la t de Student el valor calculado resultó menor que el tabulado, para una probabilidad de 0,05, lo cual demuestra que no existen diferencias significativas entre las medias alcanzadas, con un nivel de significación de un 5 %.
Los valores de % de recobro estuvieron dentro de los límites establecidos para los métodos cromatográficos (98-102 %), y los valores del coeficiente de variación para cada uno de los niveles de concentración estudiados resultaron ser menor que el 2 %, límites establecidos en la literatura para la evaluación de estos parámetros4,9 (tabla 2). En la influencia del factor concentración sobre la variabilidad de los resultados de la exactitud al aplicar la prueba de Cochran, se obtuvo que G calculada fue menor que G tabulada para una probabilidad de 0,05, k= 3 y n= 3; por lo tanto, las varianzas de las concentraciones empleadas son equivalentes, lo cual indica que la concentración no influye en la variabilidad de estos. Al realizar la prueba de significación entre la recuperación media y el 100,0 % de recuperación, con un coeficiente de variación de 0,42 %, se obtuvo una t calculada menor que t tabulada. Los resultados obtenidos demuestran la capacidad del método para dar resultados cercanos al valor verdadero, y se observa una buena exactitud.
Validación del método analítico para el ensayo de disolución
Los resultados que se obtienen en el estudio de especificidad, permiten afirmar que el método es específico en relación con los excipientes que componen la tableta porque no se observa interferencia de estos en la zona de elución del principio activo.
La curva de calibración se comportó de forma lineal en el intervalo de concentraciones comprendidas, con un adecuado valor del coeficiente de correlación, lo que demuestra la relación entre las variables concentración y respuesta detectada por el equipo empleado. El valor del coeficiente de variación de los factores de respuesta obtenido se encuentra dentro de los límites establecidos (CVf £ 5 %); la variabilidad en la relación respuesta y concentración para cada nivel evaluado es aceptada, lo que demuestra una adecuada linealidad.11
Los resultados del estudio de la precisión (tabla 3) muestran que el método es preciso ya que en todos los casos no existieron diferencias significativas entre las medias obtenidas para cada una de las muestras analizadas. Los resultados para cada uno de los valores medios obtenidos, se encuentran dentro de los límites establecidos para el estudio de precisión. Además, los valores obtenidos en las muestras evaluadas satisfacieron el límite establecido para el ensayo de disolución de las tabletas de risperidona (Q ³ 80 %).
Los resultados del estudio de la influencia de la filtración demostraron que no existieron diferencias significativas entre los resultados alcanzados, con muestras filtradas y muestras centrifugadas, ya que el valor de t calculada resultó ser menor que la t tabulada. Los resultados se encuentran dentro de los límites establecidos para el estudio de este parámetro en la validación de un ensayo de disolución.13,14 Esto permitió asegurar que la filtración no adsorbe el principio activo ni aporta interferencias al filtrado, por lo que es posible su empleo para la cuantificación de risperidona en tabletas de liberación inmediata, y se propone la filtración por papel de filtro, como medio de filtración.
El principio activo resultó estable en las condiciones del ensayo, ya que los valores del intercepto y de la pendiente después del tratamiento a 37 °C durante 1 h no difieren estadísticamente con respecto a las mediciones iniciales, pues se encuentran comprendidos en los intervalos de confianza respectivos de la curva de calibración inicial (condición necesaria y suficiente para demostrar la estabilidad del principio activo).
Comparación estadística
Al analizar estadísticamente los resultados obtenidos de los métodos analíticos por la USP 30 y el alternativo desarrollado para la cuantificación y ensayo de disolución de las tabletas de risperidona 3 mg se pudo comprobar que no existen diferencias significativas entre los resultados obtenidos por los métodos evaluados, al ser las t cal menores que la t tab. Los resultados obtenidos demuestran que es posible el empleo del método alternativo desarrollado en sustitución del método reportado en la USP para las tabletas de risperidona 3 mg.
En conclusión, el método analítico desarrollado por CLAR para la cuantificación del principio activo y el ensayo de disolución de las tabletas de risperidona 3 mg, para el control de calidad y el estudio de estabilidad, resultó ser lineal, preciso, exacto y específico en el intervalo de concentraciones establecido.
Se puede emplear el método desarrollado en sustitución del reportado en la USP 30, 2007, porque no existen diferencias significativas entre los resultados obtenidos por ambos métodos.
REFERENCIAS BIBLIOGRÁFICAS
1. Rosenstein E. Diccionario de Especialidades Farmacéuticas. 47 ed. México DF: Ed. PLM; 2001. p. 326-32.
2. British National Formulary. British Medical Association, Royal Pharmaceutical Society of Great Britain. No 27. London: BMA, RPS; 1995.
3. Martindale. The Complete Drug Referente. 32 ed. London: Pharmaceutical Press; 1999. p.1234-5.
4. Farmacopea de los Estados Unidos. USP 30 ed. The United States Pharmacopeial Convention. Estados Unidos de América NF- 25. 30 ed. Rockville: Mack Printing; 2007.
5. Quattrocchi OA, Laba RF. Introducción a la HPLC en Aplicación y práctica. Buenos Aires: Ed. Artes Gráficas Farro SA; 1992. p. 106-22, 284, 302-28.
6. Dierksneier G. Métodos cromatograficos. La Habana: Ed. Científico-Técnica; 2005. p. 1-4, 256-412.
7. Murthy KS, Ghebre-Sellasie I. Current perspectives on the dissolution stability of solid oral dosage forms. J Pharm Sci. 1993;82(2):113-26.
8. Dressman JB, Lennernas H. Oral drug absorption: prediction and assessment. New York: Marcel Dekker; 2000. p. 197-8.
9. Validation of Analytical Procedures. Technical Requirements For The Registration of Pharmaceuticals For Human Use en International Conference on Harmonization, ICH-Q2A, Geneva. 1995.
10. Fortunado D. Dissolution Technologies: Dissolution Method Development for Inmediate Release Solid Oral Dosage Forms. London: Dissolution Technologies Magazine. 2005. p.12-4.
11. Hanson WA. Handbook of Dissolution Testing. 2nd ed. Aster Publishing Corporation, Eugene, Oregon. 2002. p. 27-43, 91-113.
12. Dressman J. Oral Drugs Absorption. Aster Publishing Corporation, Eugene, Oregon. Drugs Pharm Sci. 2002;106;12-425.
13. Cox DC. Guidelines for Dissolution Testing. Aster Publishing Corporation, Eugene, Oregon. J Technol. 2001;2;41-58.
14. Skoug JW. Estrategia para el desarrollo y validación de pruebas de disolución para formas sólidas orales. New York: Pharmaceutical Technology Magazine; 1996. p. 8-15.
Recibido: 16 de julio de 2009.
Aprobado: 20 de agosto de 2009.
Lic. Caridad Margarita García Peña. Centro de Investigación y Desarrollo de Medicamentos (CIDEM). Ave. 26 No. 1605 entre Boyeros y Puentes Grandes, municipio Plaza de la Revolución, CP. 10 600, La Habana, Cuba. Correo electrónico: caridadgp@infomed.sld.cu

