










My SciELO
 Custom services
Custom servicesServices on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista Cubana de Farmacia
Print version ISSN 0034-7515On-line version ISSN 1561-2988
Rev Cubana Farm vol.44 no.3 Ciudad de la Habana July-Sept. 2010
ARTÍCULOS ORIGINALES
Desarrollo tecnológico de un antiviral de amplio espectro para administración parenteral: ribavirina 100 mg/mL
Technological development of a wide spectrum antiviral agent to parenteral administration: 100 mg/mL Ribavirin
Anna Karelia Collado CoelloI; Martha Gómez CarrilII; Odalys María Moreno LópezIII; Caridad Margarita García PeñaI; Alicia Lagarto ParraII; María Aurora BarriosIV
IMáster en Tecnología y Control de Medicamentos. Investigadora Agregada. Centro de Investigación y Desarrollo de Medicamentos (CIDEM). La Habana, Cuba.
IIMáster en Tecnología y Control de Medicamentos. Investigadora Auxiliar. CIDEM. La Habana, Cuba.
IIILicenciada en Ciencias Farmacéuticas. CIDEM. La Habana, Cuba.
IVDoctora en Ciencias. Instituto de Farmacia y Alimentos (IFAL). La Habana, Cuba.
RESUMEN
Se describe el desarrollo de una formulación que contiene ribavirina como principio activo, a una concentración de 100 mg/mL que mantiene sus propiedades estables desde el punto de vista físico, químico y microbiológico. Se realizaron los estudios de formulación correspondientes con el objetivo de determinar la formulación idónea, el procedimiento tecnológico y el envase adecuado para garantizar la estabilidad del producto terminado. Se elaboraron 3 lotes del medicamento, los cuales se envasaron en bulbos incoloros de vidrio, con calidad hidrolítica I y se almacenaron a temperatura ambiente de 30 ± 2 °C durante 12 meses; se estudió su estabilidad física y química por el método acelerado y de vida de estante. Se comprobó su estabilidad microbiológica a través de un ensayo de esterilidad, a cada uno de los lotes elaborados, al inicio y final del estudio, según la Farmacopea de los Estados Unidos 30 y la regulación establecida por el Centro Estatal de Control de Medicamentos de Cuba; se obtuvieron resultados satisfactorios. Se realizó un estudio toxicológico del inyectable que mostró un amplio margen de seguridad para ser usado en humanos. Todos los resultados obtenidos cumplieron con los límites de calidad establecidos en la literatura oficial para este tipo de forma farmacéutica, por lo que se llegó a la conclusión que el medicamento desarrollado está correctamente formulado desde el punto de vista galénico con un tiempo de vida útil de 12 meses, almacenado bajo las condiciones estudiadas. Finalmente el medicamento fue introducido al nivel industrial sin que se presentaran problemas tecnológicos.
Palabras clave: Desarrollo tecnológico, antiviral, solución inyectable, ribavirina.
ABSTRACT
Development of a formula containing Ribavirin as active principle at a 100 mg/mL concentration maintaining its stable properties from the physical, chemical and microbiological point of view is described. The aim of present formula studies was to determine the suitable formula, the technological procedure and the appropriate package to guarantee stability of end product. Three drugs batches were manufactured, which were packing in colourless glass bulbs with a hydrolytic I quality and stored at room temperature of 30 ± 2 ºC for 12 months; its physical and chemical was studied by accelerated method and shelf life. Its microbiological stability was verified by sterility trial in each of processed batches at study onset and at the end according to USA Pharmacopeia-30 and the regulation established by Cuban State Center for Drug Control with satisfactory results. A toxicology study was conducted of injectable agent showing a wide safety margin to human use. All results obtained fulfilled the quality limits established in official literature for this type of pharmaceutical way thus concluded that the developed drug is properly formulated from the medical point of view with a useful life time of 12 months, stored under study conditions. Finally, the drug was introduced at industrial level without technological problems.
Key words: Technological development, antiviral, injectable solution, Ribavirin.
INTRODUCCIÓN
La ribavirina es un análogo de nucleósido que estructural y funcionalmente se asemeja a la guanosina. Es activa contra una amplia gama de virus DNA y RNA in vitro, pero tiene pobre actividad in vivo. Numerosos mecanismos de acción han sido propuestos; en su forma monofosfatada, la ribavirina es un inhibidor competitivo potente de la enzima dehidrogenasa que es esencial para la síntesis de trifosfato de guanosina.1,2
Esta inhibición resulta en una disminución en los depósitos celulares de guanidina, necesaria tanto para la multiplicación viral como celular. Sin embargo, su mayor efecto antiviral de significación es la inhibición en la terminación o punto final del RNA mensajero viral y produce una caída en la producción de proteínas virales.3 También ha sido autorizado su empleo en niños con bronquiolitis severa debidas al virus sincitial respiratorio.4
Con el objetivo de sustituir medicamentos por concepto de importaciones al país, es que en el Centro de Investigación y Desarrollo de Medicamentos (CIDEM) se hace necesario el desarrollo del inyectable de ribavirina 100 mg/mL de producción nacional.
Por tanto, el objetivo fundamental de este trabajo, consistió en desarrollar una solución inyectable de ribavirina, que brindara una dosis de 100 mg por cada mililitro de preparación, cumpliera con todos los requerimientos para esta forma farmacéutica y proporcionara el efecto terapéutico deseado.
Como objetivos específicos para lograr lo anteriormente planteado, se propuso comprobar la calidad farmacéutica, tanto del principio activo como de las sustancias auxiliares utilizadas en su composición; realizar los ensayos de preformulación necesarios para la posterior selección de la formulación más prometedora así como la selección del envase adecuado; desarrollar un método para la determinación de ribavirina por cromatografía líquida de alta resolución (CLAR) para el control de calidad y los estudios de estabilidad del producto terminado, además de estudiar la estabilidad de la formulación de ribavirina 100 mg/mL. Finalmente, presentar el expediente de registro del medicamento en el Centro Estatal de Control de Medicamentos de Cuba (CECMED) para que una vez aprobado, pueda aumentar el arsenal terapéutico de nuestro país.
MÉTODOS
Desarrollo tecnológico
En el desarrollo tecnológico de la formulación, se empleó ribavirina materia prima, correspondiente al lote 3200773001, del fabricante Chemo y fue importada por FARMACUBA desde la India, valorada previamente en los laboratorios del CIDEM, realizándose los ensayos establecidos en las especificaciones de calidad reportadas en la USP 30.5
Para el análisis del principio activo se empleó un patrón de ribavirina USP con un nivel de pureza de 0,0997 mg/mg. Las sustancias auxiliares utilizadas se analizaron por las técnicas oficiales correspondientes (USP 30), para comprobar la concordancia con los requisitos exigidos para cada una de ellas. Todos los reactivos empleados son de calidad para análisis y los equipos y cristalería de laboratorio se encontraban debidamente certificados. Para el envase se utilizaron bulbos de vidrio incoloro, de 6 mL de capacidad, clase hidrolítica I, cierre hermético que también fueron analizadas para comprobar que cumplían con los requisitos establecidos para su uso farmacéutico.
Se realizaron 5 ensayos tecnológicos en los que se ajustó el pH y la isotonicidad según las exigencias de un preparado parenteral, con el objetivo de obtener la formulación más adecuada. Todas las formulaciones ensayadas presentaban ribavirina como principio activo y agua para inyección csp (1 mL) y se diferenciaban en los excipientes. La formulación 1 presentaba cloruro de sodio; la 2 cloruro de sodio y citrato de sodio; la 3 ácido cítrico monohidratado e hidróxido de sodio; la 4 citrato de sodio, ácido cítrico monohidratado e hidróxido de sodio y la 5 citrato de sodio y ácido cítrico monohidratado.
La isotonicidad se ajustó con cloruro de sodio haciendo el cálculo teórico por el método del equivalente de cloruro de sodio. Se evaluó el proceso de esterilización bajo presión de vapor y por membrana esterilizante. Para evaluar la compatibilidad de la membrana con las 5 soluciones objeto de ensayo, se colocaron membranas de nitrato de celulosa, acetato de celulosa y celulosa regenerada, de 47 mm de diámetro, en placas Petri y se cubrieron con la solución, manteniéndolas en contacto durante 24 h. Transcurrido este tiempo se realizó un análisis microscópico y macroscópico de las membranas así como el contenido de principio activo, comparándolas con una muestra sin filtrar.6
Muestras de los 5 ensayos se conservaron a temperatura ambiente (30 ± 2 °C) y a temperatura de 40 °C durante 2 meses y se les estudió el comportamiento en cuanto a sus características organolépticas, pH, contenido de principio activo y los productos de degrada
ción.7
Con la fórmula y técnica de cada uno de estos ensayos se elaboraron dos, de 1 L ada uno y se dividieron en 2 porciones para evaluar el proceso de esterilización final: presión de vapor a 121 °C. Muestras de estos ensayos se conservaron a 60 °C y se observaron durante 2 meses con el objetivo de definir la formulación a proponer, evaluándose en el tiempo las características organolépticas, pH y el contenido de principio activo.
Una vez propuesta la formulación, con los diferentes parámetros de trabajo establecidos, se procedió a la elaboración de 3 lotes pilotos de 5 000 mL de volumen total cada uno, siguiendo el método de fabricación propuesto en la figura para realizar los estudios de estabilidad del producto final, y definir sus condiciones de almacenamiento. Las muestras fueron analizadas recién elaboradas y transcurrido 1 año a diferentes temperaturas.8
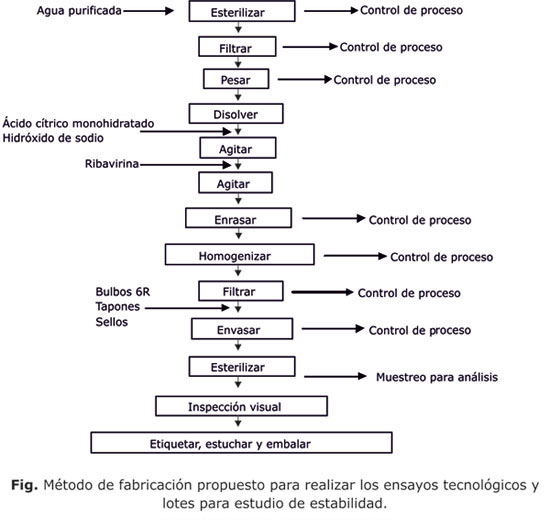
Desarrollo de la técnica de análisis para el control de la calidad y estudio de estabilidad del producto terminado
Se desarrolló un método de análisis por CLAR para la cuantificación de ribavirina en el inyectable de 100 mg/mL, por no existir un método reportado en las farmacopeas oficiales. La validación fue realizada según la Categoría I (USP-30) de Medicamentos y la Regulación 41-2007 (CECMED), evaluándose los parámetros que a continuación se describen: linealidad, precisión, especificidad y exactitud. El control de la calidad del producto terminado se realizó por CLAR, empleando para el análisis el método reportado en la USP.
Estudio de estabilidad
El presente estudio se realizó por el método de estabilidad acelerada y de vida de estante. Se emplearon muestras de 3 lotes identificados como: 07001, 07002 y 07003, envasados en bulbos de vidrio incoloro formato 6R, CH-I, con tapón de goma butílica para no liofilizados y 20 mm de diámetro, además del sello de aluminio anodizado.
Para el estudio de estabilidad acelerada, se almacenaron muestras de los lotes en una estufa con temperatura controlada de 40 °C y 75 % de humedad relativa (HR) y se valoraron al inicio, 2, 3 y 6 meses de elaborados; se evaluaron en el período evaluado las características organolépticas, el pH, el contenido de principio activo, así como la presencia de los productos de degradación teniendo en cuenta los resultados obtenidos en el estudio de especificidad del método cromatográfico desarrollado y validado al someter la muestra a condiciones drásticas.
Para el estudio de estabilidad por vida de estante, los lotes estudiados fueron almacenados a temperatura ambiente (30 ± 2 °C y HR de 70 ± 5 %), y se valoraron al inicio, a los 3, 6, 9 y 12 meses de fabricados.9
Para el estudio toxicológico se empleó la determinación de la toxicidad aguda endovenosa para lo cual se utilizaron ratones albinos Suizos hembras procedentes de la colonia de la UCTB Control Biológico. Se ensayaron 4 niveles de dosis con el objetivo de obtener una relación dosis-efecto y determinar la DL50.
Se realizó una prueba microbiológica a la formulación empleando el método de filtración por membrana, reportado en la USP,5 y se analizó al inicio y a los 12 meses de comenzado el estudio.
RESULTADOS
Se comprobó que todas las sustancias, así como el material de envase utilizado en el estudio realizado, cumplieron con los requerimientos correspondientes para considerarlos aptos para uso farmacéutico.
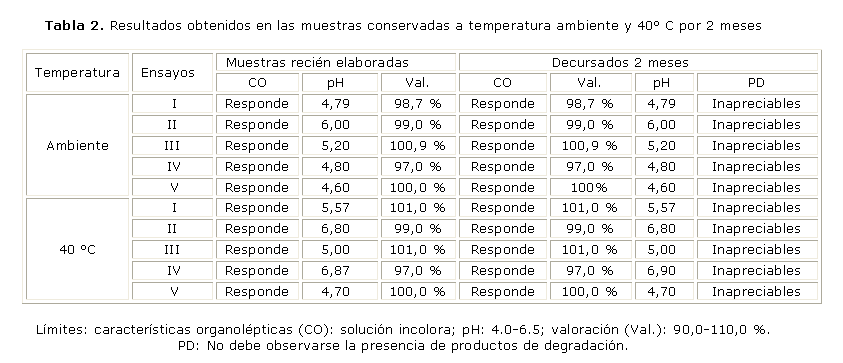
En la tabla 1 se muestran los resultados del estudio realizado para determinar la compatibilidad de las membranas. Mientras que en la tabla 2, se puede observar el comportamiento de las muestras durante 2 meses conservadas a temperatura ambiente y a 40 °C.
Tabla 1. Resultados obtenidos con los diferentes medios de filtración a tiempo= 0 y t= 24 h
| Ensayos/parámetros | t= 0 | t= 24 h | |||||
| NC | CR | AC | NC | CR | AC | ||
| I | CO | - | - | - | Integra | Integra | Integra |
| Val. | 98,7 % | 98,8% | 99,0% | 99,2% | 99,2% | 99,4% | |
| pH | 4,79 | 4,77 | 4,80 | 4,80 | 4,82 | 4,80 | |
| II | CO | - | - | - | Integra | Integra | Integra |
| Val. | 99,0 % | 99,0% | 99,0% | 99,7% | 99,7% | 99,5% | |
| pH | 6,00 | 6,22 | 6,00 | 6,45 | 6,45 | 6,80 | |
| III | CO | - | - | - | Integra | Integra | Integra |
| Val. | 100,9 % | 100,9 % | 100,9 % | 101,0% | 101,0% | 101,0% | |
| pH | 5,20 | 5,20 | 5,20 | 5,22 | 5,22 | 5,22 | |
| IV | CO | - | - | - | Integra | Integra | Integra |
| Val. | 97,0 % | 97,0 % | 97,0 % | 97,6% | 97,5% | 97,0% | |
| pH | 4,80 | 4,81 | 4,87 | 5,57 | 5,80 | 5,70 | |
| V | CO | - | - | - | Integra | Integra | Integra |
| Val. | 100,0% | 100,2 % | 100,2 % | 100,0% | 101,0% | 100,1% | |
| pH | 4,60 | 4,65 | 4,56 | 4,70 | 4,70 | 4,65 | |
| Ss/f | CO | - | - | - | Integra | Integra | Integra |
| Val. | 101,5 % | 101,0 % | 101,3 % | 101,3% | 101,7% | 101,8% | |
| pH | 4,73 | 4,77 | 4,76 | 4,76 | 4,76 | 4,78 | |
Límites: características organolépticas (CO): solución incolora; pH: 4,0-6,5;
Valoración (Val.): 90,0-110,0 %; análisis de la membrana: membrana que no se afecta por ninguno de los componentes de la formulación; Ss/f: solución sin filtrar; NC: nitrato de celulosa; AC: acetato de celulosa; CR: celulosa regenerada.
Se desecharon las formulaciones que no cumplieron con el parámetro pH y se dirigió el trabajo con las 2 formulaciones resultantes con las que se elaboraron 2 ensayos de 1 L cada uno para evaluar además si es factible la esterilización final a 121 °C, conservadas entonces a temperatura ambiente y a 60 °C. Esos resultados se muestran en la tabla 3.
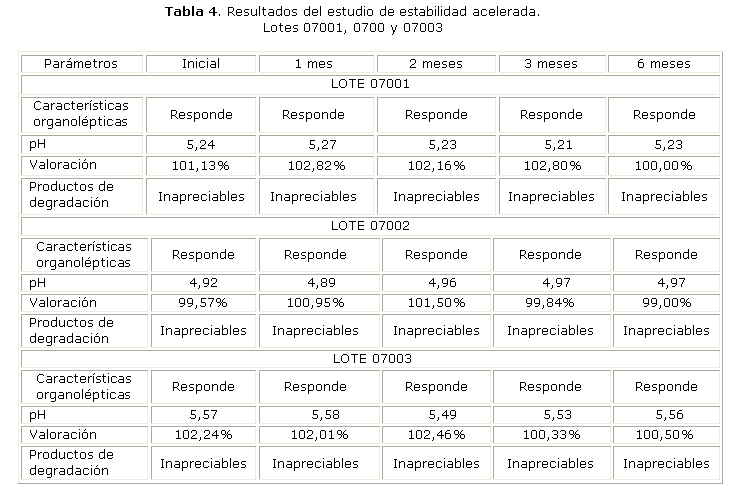
Los resultados del estudio de estabilidad acelerada a 40 °C y 75 % de HR, se reportan en la tabla 4; se demostró que el producto terminado no presentó cambios significativos.
Los resultados del estudio de vida de estante, al inicio, 3, 6, 9 y 12 meses, se muestran en la tabla 5; se observó que el producto terminado cumplió con las especificaciones de calidad durante el estudio.
Tabla 5. Resultados del estudio de estabilidad por vida de estante.
Lotes 07001,07002 y 07003
| Parámetros | Inicial | 3 meses | 6 meses | 9 meses | 12 meses |
| LOTE 07001 | |||||
| Características organolépticas | Responde | Responde | Responde | Responde | Responde |
| pH | 5,24 | 5,19 | 5,2 | 5,2 | 5,2 |
| Valoración | 101,13% | 102,50% | 99,90% | 101,00% | 100,10% |
| Productos de degradación | Inapreciables | Inapreciables | Inapreciables | Inapreciables | Inapreciables |
| Esterilidad | Cumple | - | - | - | Cumple |
| LOTE 07002 | |||||
| Características organolépticas | Responde | Responde | Responde | Responde | Responde |
| pH | 4,92 | 4,96 | 4,86 | 4,87 | 4,86 |
| Valoración | 99,57% | 99,90% | 98,60% | 99,80% | 99,20% |
| Productos de degradación | Inapreciables | Inapreciables | Inapreciables | Inapreciables | Inapreciables |
| Esterilidad | Cumple | - | - | - | Cumple |
| LOTE 07003 | |||||
| Características organolépticas | Responde | Responde | Responde | Responde | Responde |
| pH | 5,57 | 5,55 | 5,56 | 5,55 | 5,57 |
| Valoración | 102,24% | 99,39% | 101,90% | 99,10% | 101,00% |
| Productos de degradación | Inapreciables | Inapreciables | Inapreciables | Inapreciables | Inapreciables |
| Esterilidad | Cumple | - | - | - | Cumple |
DISCUSIÓN
Desarrollo tecnológico
En la evaluación de las muestras conservadas a temperatura ambiente (30 ± 2 °C) y 40 °C por 2 meses puede apreciarse que las formulaciones responden a las características organolépticas, y al contenido de principio activo tanto a temperatura ambiente como a 40 °C no se observaron cambios en su coloración y la disminución del principio activo fue inferior al 5 %, límite establecido por nuestro órgano regulador, no siendo así en el parámetro pH donde las únicas formulaciones que cumplen son la III y la V, ya que mantuvieron sus valores dentro de los límites establecidos, observándose en este último parámetro evaluado una disminución significativa.
La evaluación de las dos variantes resultantes del estudio anterior, en cuanto al aumento de temperatura arrojó que el aumento de la temperatura no provoca cambios significativos en las dos formulaciones estudiadas, así como no se observan cambios cuando las muestras se someten a esterilización final por presión de vapor, indicativo de que pueden ser empleadas siguiendo el método de fabricación propuesto. En nuestro trabajo se decidió escoger la fórmula III ya que desde el punto de vista económico los excipientes empleados son menos costosos que los empleados en la formula V.
Lotes para estudio de estabilidad
Con la fórmula y procedimiento propuesto se elaboraron 3 lotes de 5 L cada uno, identificados como 07001, 07002 y 07003, en los que se pudo comprobar que a nivel piloto la tecnología desarrollada se comporta correctamente lográndose un producto que cumple con las especificaciones de calidad requeridas, los que fueron sometidos a diferentes tratamientos de temperatura para evaluar el tiempo de vida útil del medicamento.
Estudio de estabilidad
La validación de la técnica de análisis para la determinación del principio activo mostró que esta es específica, precisa y que cumple con los criterios de linealidad y exactitud, por lo que pudo ser utilizada para el estudio de estabilidad del medicamento.
Los resultados del estudio de estabilidad acelerada, se reportan en la tabla 4; se encontró que en las condiciones establecidas (temperatura controlada 40 °C y 75 % de HR se mantiene la estabilidad del producto, ya que el tratamiento no alteró su calidad final, y se mantienen las características organolépticas, pH y valoración dentro de los límites establecidos en las especificaciones de calidad.
Según los valores obtenidos en el estudio de estabilidad por vida estante que se muestran en la tabla 5, se puede concluir que el producto presenta una estabilidad satisfactoria del principio activo a temperatura ambiente (30 ± 2 °C), ya que se mantienen los parámetros que determinan su calidad en los 12 meses de fabricado el producto; no se observaron variaciones notables entre la valoración inicial y la obtenida transcurrido todo el tiempo de estudio. Los valores de pH se mantuvieron dentro de los límites permisibles para esta formulación. En cuanto a las características organolépticas, estas permanecieron invariables durante este período.
De igual manera se comportaron los resultados de microbiología; se pudo comprobar que no aparece crecimiento microbiano, transcurridos 12 meses de elaborado el producto.
El estudio toxicológico concluyó que el inyectable provocó una DL50 de 1 734,38 mg/kg clasificándose en la categoría 4 según el Sistema Armonizado de la OECD, o sea, que su margen de seguridad es amplio.
Por todos estos resultados obtenidos, se proponen 12 meses como fecha de vencimiento para la formulación estudiada, almacenada a temperatura ambiente (30 ± 2 °C) y envasados en bulbos de vidrio incoloro, de 6 mL de capacidad, clase hidrolítica I con cierre hermético.
REFERENCIAS BIBLIOGRÁFICAS
1. Pérez Ávila J, Ramos García A. Antivirales. Consideraciones actuales. Rev Acta Médica. 1990;4(2):268-83.
2. Hall CB, Mc Bride JT, Gala CL. Ribavirin greatment of respiratory syncytial virus infection in infants with underlying cardiopulmonary disease. JAMA. 1985;254:3047.
3. Shepp DH, Dandliker PS, Meyers JD. Treatment of varicella-zoster virus infection in severely inmunocompromised patients. N Engl J Med Magazine. 1986;314:208.
4. Lundgren G, Wilczek H, Lonnqvist B. Acyclovir prophylaxis in bone marrow trasplant receipients. Scand J Infect Dis.1985;47(Suppl):137.
5. United States Pharmacopoeial Convention. USP 30. United States Pharmacopoeia 30 and National Formulary No 25. Rockville: Mack Printing; 2007.
6. Voigt R. Tratado de tecnología farmacéutica. Zaragoza: Editorial Acribia; 1979. p. 417, 427, 430, 442-443, 631, 540.
7. Diccionario de Especialidades Farmacéuticas. 50 ed. Madrid: Reverté; 2004. p. 329-347, 675-717.
8. Akers MJ. Parenteral Quality Control. 2nd ed. New York: American Chemical Society; 1994.
9. Sigarroa A. Biometría y diseño experimental. La Habana: Ed. Pueblo y Educación; 1985. p. 430.
Recibido: 8 de abril de 2010.
Aprobado: 17 de mayo de 2010.
M. C. Anna Karelia Collado Coello. Centro de Investigación y Desarrollo de Medicamentos (CIDEM). Ave. 26 No. 1 605 entre Boyeros y Puentes Grandes. CP 10 600. Plaza de la Revolución, La Habana, Cuba.


{kind=link}
{kind=link}
{kind=link}