










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Farmacia
versión impresa ISSN 0034-7515versión On-line ISSN 1561-2988
Rev Cubana Farm v.44 n.3 Ciudad de la Habana jul.-sep. 2010
ARTÍCULOS ORIGINALES
Validación del método analítico aplicable al estudio de estabilidad de risperidona solución oral 1 mg/mL
Validation of an analytical method applicable to study of 1 mg/mL oral Risperidone solution stability
Maikel Abreu ÁlvarezI; Caridad Margarita García PeñaII; Lissette Martínez MirandaIII; Adriana Muñoz CernadaII; Mirna Fernández CerveraIV; Vivian Martínez EspinosaV
IMáster en Tecnología y Control de Medicamentos. Empresa Farmacéutica "Juan R. Franco". La Habana, Cuba.
IIMáster en Tecnología y Control de Medicamentos. Investigadora Agregada. Centro de Investigación y Desarrollo de Medicamentos (CIDEM). La Habana, Cuba.
IIILicenciada en Bioquímica. Investigadora Auxiliar. CIDEM. La Habana, Cuba.
IVDoctora en Ciencias Farmaceuticas. Instituto de Farmacia y Alimentos (IFAL). La Habana, Cuba.
VTécnico Medio en Química. CIDEM. La Habana, Cuba.
RESUMEN
Se validó un método analítico por cromatografía líquida de alta resolución, aplicable al estudio de estabilidad de risperidona solución oral 1 mg/mL. El método analítico validado resultó lineal, preciso, específico y exacto. Se desarrolló el estudio de estabilidad de la solución oral de risperidona 1 mg/mL y se determinó su fecha de vencimiento. El estudio de vida de estante se desarrolló por un periodo de 24 meses a temperatura ambiente; mientras que el estudio de estabilidad acelerada se efectuó sometiendo el producto a la influencia de la humedad y la temperatura; se realizó el análisis durante 3 meses. La formulación cumplió con las especificaciones de calidad descritas en la Farmacopea. Los resultados del estudio de estabilidad por vida de estante después de transcurridos los 24 meses indicaron que el producto mantiene los parámetros que determinan su calidad durante ese tiempo, y en los estudios acelerados no se observó degradación significativa (p> 0,05) del producto. Se estableció 2 años como fecha de vencimiento en las condiciones señaladas.
Palabras clave: Risperidona, validación, estudios de estabilidad.
ABSTRACT
A validated analytical method by high-performance liquid chromatography (HPLC) was applicable to study of 1 mg/mL Risperidone oral solution stability. The above method was linear, accurate, specific and exact. A stability study of the 1 mg/mL Risperidone oral solution was developed determining its expiry date. The shelf life study was conducted for 24 months at room temperature; whereas the accelerated stability study was conducted with product under influence of humidity and temperature; analysis was made during 3 months. Formula fulfilled the quality specifications described in Pharmacopeia. The results of stability according to shelf life after 24 months showed that the product maintains the parameters determining its quality during this time and in accelerated studies there was not significant degradation (p> 0.05) in the product. Under mentioned conditions expiry date was of 2 years.
Key words: Risperidone, validation, stability studies.
INTRODUCCIÓN
Risperidona es un antipsicótico benzisoxazólico, con efectos antagonistas de receptores de dopamina D2 y serotonina (5-HT2), adrenérgicos (a1 y a2) e histamina (H1). Es un antipsicótico atípico. La solución oral de risperidona 1 mg/mL se indica en el tratamiento de la psicosis crónica y aguda; manía.1,2
La cromatografía líquida de alta resolución (CLAR) le brinda la posibilidad al analista de emplear esta herramienta para solucionar los inconvenientes de los métodos espectrofotométricos en los estudios de estabilidad, pues además de presentar una alta sensibilidad y exactitud, es en esencia un método separativo; lo que permite medir con gran selectividad el compuesto deseado, siempre y cuando se encuentre un sistema cromatográfico que asegure una adecuada separación.3,4
La validación es el proceso establecido para la obtención de pruebas documentadas de que el método es lo suficientemente fiable para producir el resultado esperado. Los parámetros analíticos que pueden ser considerados en la validación de un método analítico, según se expresa en la United States Pharmacopoeia (USP 30), son: exactitud, precisión, especificidad, límite de detección, límite de cuantificación, linealidad, rango, tolerancia y robustez.5,6
La estabilidad de los productos farmacéuticos representa un importante eslabón en el desarrollo y formulación de toda forma terminada. De esta manera se puede definir las condiciones de almacenamiento en el envase propuesto y establecer la vida útil del producto farmacéutico. Estos estudios contemplan la conservación de la potencia, pureza, características organolépticas y su efectividad.7
El Centro para el Control Estatal de los Medicamentos de la República de Cuba (CECMED) exige para el registro de un nuevo medicamento la realización de los estudios de estabilidad. En el presente trabajo se empleó un método analítico validado en el Centro de Investigación y Desarrollo de Medicamentos (CIDEM) para el estudio de estabilidad del producto terminado.
La presente investigación tuvo como objetivo validar el método de análisis para el estudio de estabilidad del producto terminado y realizar el estudio de estabilidad de una formulación de risperidona solución oral 1 mg/mL y determinar su fecha de vencimiento.
MÉTODOS
La sustancia de referencia química de risperidona fue suministrada por el grupo de sustancias de referencia del CIDEM la cual fue analizada por el método cromatográfico establecido para realizar el control de la calidad de la materia prima, con una pureza de 99,8 %. El producto terminado en forma de solución oral, fue elaborado en el Laboratorio de Formas Terminadas del CIDEM, identificado como los lotes 6001, 6002 y 6003, fabricado en marzo de 2006, con fecha de vencimiento marzo de 2008, los cuales cumplieron con las especificaciones de calidad establecidas para el control de la calidad.
Todos los reactivos utilizados fueron de grado HPLC, de la Merck, Alemania. En el ensayo se empleó un cromatógrafo (KNAUER) con detector UV/VIS (KNAUER) ajustado a 278 nm, un inyector con un loop de 20 µL e integrador (SHIMADZU CR 8 A). La separación se realizó isocráticamente empleando una columna Lichrosorb RP-18 (5 µm) (250 x 4 mm) y un flujo de 1,5 mL/min. La fase móvil consistió en una mezcla desgasificada del solución amortiguadora fosfato de potasio 0,05 Molar a pH= 4,6, y acetonitrilo en una proporción de 55:45.
La solución de referencia se obtuvo disolviendo risperidona, sustancia de referencia, en ácido clorhídrico hasta lograr una concentración final de 100 µg/mL. En la preparación de la solución de ensayo para la valoración, se disolvió una alícuota de la muestra de la solución oral ácido clorhídrico para obtener una concentración final de 100 µg/mL
Validación del método analítico
La validación fue realizada según la categoría I (USP 30) empleando el método reportado en USP 30; se evaluaron los parámetros que a continuación se describen.
Para evaluar la especificidad del método cromatográfico utilizado se emplearon muestras de producto terminado y una muestra que contenía solo los exipientes (ácido benzoico, ácido tartárico, hidróxido de sodio, sacarina sódica, aceite esencial de naranja dulce, talco, alcohol etílico clase C, agua desionizada c.s.), las cuales se sometieron a condiciones extremas de temperatura (40 ºC) a pH ácido (ácido clorhídrico 1 M) y alcalino (hidróxido de sodio 1 N), además de medio oxidante (con peróxido de hidrógeno). Criterio de aceptación: No se debían obtener señales del placebo y de los productos de degradación en la zona de elusión del principio activo. Las áreas bajo las curvas del patrón y de la risperidona en el producto terminado debían ser similares.
Para la linealidad se realizó una curva de calibración que compredió las concentraciones en el intervalo 20 y 200 µg/mL, se prepararon muestras con diferentes concentraciones de risperidona que representaron el 20, 50, 80, 100, 150 y 200 % de la concentración teórica del principio activo en la solución oral. Se determinó la ecuación de la recta, el coeficiente de correlación, la prueba de significación estadística de significación de la pendiente Sb rel (%), los coeficiente de variación de los factores de respuesta y el ensayo de proporcionalidad.
Se realizó el cálculo de la precisión intermedia del método. La repetibilidad se hizo sobre la base de 10 determinaciones y se estudió la precisión intermedia haciendo las valoraciones por 2 analistas y en 3 diferentes días a 3 valores de concentración, con ellas se determinaron los valores medios, la desviación estándar y el coeficiente de variación. La precisión intermedia se determinó utilizando muestras de un lote de producto terminado. Se cuantificaron por el método cromatográfico propuesto y se determinaron mediante el procesamiento estadístico, la media, la desviación estándar, el coeficiente de variación y la existencia o no de diferencias significativas entre los analistas, los días y las tres concentraciones para los resultados de cada muestra. Para comprobar la precisión intermedia del método, se aplicaron las pruebas de Fisher y de la t Student para determinar si existían diferencias significativas entre los resultados al variar las condiciones de análisis.
Para la exactitud se prepararon soluciones que contenían los excipientes a las cuales se le adicionaron concentraciones conocidas de risperidona que correspondían al 80, 100 y 120 % de lo declarado de principio activo, según la formulación propuesta, determinándose el porcentaje de recuperación, la desviación estándar y el coeficiente de variación. Se aplicó además el ensayo de Gochran con vistas a comprobar si la variación de la concentración producía diferencias significativas en los resultados y la prueba de la t de Student para determinar diferencias significativas entre la recuperación media y el 100 %.
Estudio de estabilidad
Este estudio se realizó por los métodos de vida de estante y estabilidad acelerada. Se emplearon muestras de los lotes 06001, 06002 y 06002, producidos en el Laboratorio de Formas Líquidas del CIDEM y envasados en frasco de vidrio ámbar con tapa Pilfer Proof, calidad hidrolítica II, de 125 mL de capacidad nominal, acabado 28 mm. Tapa plástica diámetro interno 28 mm.
Estabilidad acelerada
En los estudios de estabilidad acelerada, se almacenaron las muestras de los lotes estudiados en un horno a temperatura controlada de 40 ºC y 75 % de humedad relativa; se valoraron al inicio, 1, 2, 3 y 6 meses.
Estabilidad por vida de estante
En el estudio de estabilidad por vida de estante, los lotes estudiados se almacenaron a temperatura ambiente (30 ºC) y protegidos de la luz; se valoraron al inicio, a 3, 6, 9, 12, 18 y 24 meses de fabricados. Se realizó la determinación de la esterilidad a la solución oral al inicio y al final del estudio (24 meses), empleando para ello el método general para la realización del conteo microbiológico, reportado en la USP 30, 2007.
Procesamiento estadístico de los datos
Para el procesamiento estadístico de los datos se hizo un análisis descriptivo en el cual se calcularon la media y la desviación estándar para cada muestra. Se aplicó además el ensayo de normalidad (Anderson- Darling) y el ensayo de homogeneidad de varianza de Levene. Se aplicó el ensayo de comparación múltiple de medias de Student-Newman-Keuls, para determinar las diferencias entre grupos. El nivel de significación estadística, empleado en todos los casos, fue como mínimo de p< 0,05. Los datos fueron procesados utilizando el paquete estadístico MINITAB, versión 14.0.
RESULTADOS
Validación del método analítico
La figura muestra los resultados del estudio de especificidad del método. Como se observa en el cromatograma correspondiente a la muestra de excipientes (A), no se obtuvo ninguna señal en la zona de interés, al ser comparado con la señal obtenida para las soluciones estándar de referencias (B) y de la muestra de la solución oral (C), lo cual indica que los excipientes o sustancias auxiliares de la solución no interfirieron en la determinación del principio activo. En cuanto a las muestras sometidas a condiciones drásticas de hidrólisis básica (D) e hidrólisis ácida (E), indicadas en los cromatogramas, la molécula resultó ser estable, sólo se pudo apreciar una ligera degradación por la disminución de la altura de los picos, sin la aparición de pequeños picos secundarios como posibles productos de degradación.
En la tabla 1 se reportan los resultados del estudio de la linealidad del sistema, el coeficiente de regresión lineal fue de 0,9998 y el coeficiente de variación del factor de respuesta resultó igual a 4,2 %. Los resultados del estudio de precisión del método aparecen en la tabla 2. En el estudio de repetibilidad realizado, la media obtenida fue de 100,31 % y el coeficiente de variación fue de 0,22 %, mientras que los valores de F calculadas y los valores de t calculadas fueron menores que los valores tabulados, para un 95 % de confianza, para cada uno de los niveles estudiados.
Tabla 2. Estudio de la precisión intermedia del método analítico
| Niveles (%) | Analista 2 (%) | |||||||||||
| Primer día | Segundo día | Tercer día | Primer día | Segundo día | Tercer día | |||||||
| 80 | 79,9 ± 0,2 | 80,1 ± 0,1 | 79,9 ± 0,1 | 79,9 ± 0,1 | 80,1 ± 0,1 | 79,9 ± 0,1 | ||||||
| 100 | 99,7 ± 0,2 | 99,9 ± 0,1 | 99,7 ± 0,2 | 99,9 ± 0,1 | 99,7 ± 0,1 | 99,9 ± 0,1 | ||||||
| 120 | 120,0 ± 0,1 | 120,0 ± 0,1 | 119,9 ± 0,2 | 119,9 ± 0,2 | 120,0 ± 0,2 | 119,9 ± 0,2 | ||||||
| Análisis estadístico | ||||||||||||
| Prueba de Fischer | ||||||||||||
| Niveles (%) | Prueba de significación de Fisher | Límites | ||||||||||
| por analistas ( F tab ( 8/8; 0.05) = 3,44) | por día ( F tab (5/5; 0.05) = 5,05 ) | |||||||||||
| Días ½ | Días 2/3 | Días 1/3 | exp≤ Ftab | |||||||||
| 80 | 1,47 | 1,78 | 1,36 | 1,31 | ||||||||
| 100 | 1,59 | 1,11 | 1,44 | 1,44 | ||||||||
| 120 | 1,25 | 1,27 | 2,07 | 2,06 | ||||||||
| Niveles (%) | Prueba de significación de t de Student | Límites | ||||||||||
| por analistas (ttab(16; 0,05) = 2,12) | por día ( ttab(10; 0,05)= 2,22) | |||||||||||
| Días ½ | Días 2/3 | Días 1/3 | Fexp≤ Ftab | |||||||||
| 80 | 0,38 | 1,40 | 2,01 | 0,53 | ||||||||
| 100 | 0,08 | 1,12 | 0,17 | 0,94 | ||||||||
| 120 | 0,63 | 0,57 | 0,75 | 0,30 | ||||||||
| Coeficientes de variación | ||||||||||||
| Niveles (%) | Analista 1 | Analista 2 | Límites | |||||||||
| 80 | 0,22 | 0,18 | CV≤ 2,0 % | |||||||||
| 100 | 0,24 | 0,19 | ||||||||||
| 120 | 0,14 | 0,16 | ||||||||||
tcal: t calculada; Fcal: F calculada; ttab: t tabulada; Fcal: F tabulada; %: Los valores de porcentaje de principio activo (n= 3) se representan como valor medio ± desviación estándar relativa. Niveles: porcentaje de la concentración principio activo declarado en la formulación.
En la tabla 3 aparecen los resultados del estudio de exactitud. La recuperación media fue de 99,77 % y el valor de t calculada (1,44) y de G calculada (0,5000) fueron menores que los valores tabulados, para un 95 % de confianza, t tabulada 2,306 y G tabulada 0,8709.
Estudio de estabilidad
Los resultados del estudio de estabilidad acelerada a 40 ºC y 75 % de humedad relativa (HR), correspondiente a los lotes 06001, 06002 y 06003 (tabla 4), demostraron que el producto terminado no presentó cambios en los valores de los parámetros evaluados durante el estudio.
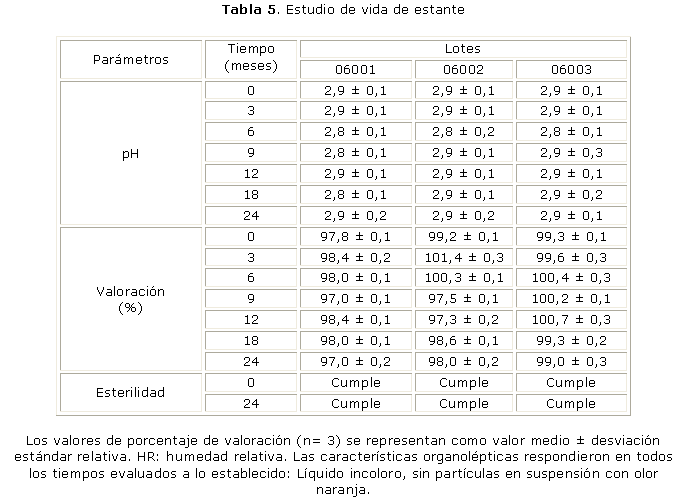
Los resultados del estudio de vida de estante, al inicio, 3, 6, 9, 12, 18 y 24 meses (tabla 5), muestran que el producto terminado cumplió con las especificaciones de calidad durante el estudio.
DISCUSIÓN
Validación del método analítico
Los resultados del estudio de especificidad del método (figura), demuestran la especificidad del método al no presentarse interferencias de picos adicionales en la zona de elución del producto principal, ya que los productos de degradación y los excipientes de la formulación presentan tiempos de retenciones diferentes al principio activo, todos inferiores al tiempo de retención del principio activo. Este estudio permitió concluir que el método cromatográfico resultó ser específico pues permite cuantificar el principio activo después de la degradación del mismo en muestras de medicamento sin interferencias de excipientes o producto de degradación. En cuanto a las muestras sometidas a condiciones drásticas no se observó la aparición de picos secundarios, atribuibles a un posible producto de degradación, ni interferencia en la determinación del principio activo, lo que demuestra la especificidad del método desarrollado.4,5
Los resultados de los estudios de linealidad mostraron coeficiente de regresión y de determinación superiores a los exigidos en la Regulación 41-2007 del CECMED, 0,99 y 0,98 respectivamente, demostrándose la existencia de correlación con una probabilidad elevada, así como el grado de relación entre las variables concentración y respuesta detectada por el equipo empleado. También el coeficiente de variación de los factores de respuesta y la desviación estándar relativa de la pendiente fueron inferiores al normado como máximo para estos indicadores: 5 y 2 %, respectivamente, ambos son considerados estimadores puntuales que permiten caracterizar la variabilidad. El valor del coeficiente de variación del factor de respuesta permitió demostrar que existía variabilidad en la relación respuesta y concentración para cada valor evaluado. El intervalo de confianza del intercepto incluyó al cero, lo que permitió excluir la significación del error del intercepto. Se demostrándose con estos resultados la linealidad del método propuesto.4-6
En los estudio de la repetibilidad realizado a una misma muestra, por el mismo analista, el mismo día, a través de 6 réplicas, se alcanzaron coeficientes de variación adecuados, lo que demostró la buena precisión del método, observándose una variabilidad de los resultados dentro de los límites establecidos para los métodos cromatográficos: C£ 2,0%.4,5
Los valores que se obtuvieron en los estudios de precisión intermedia, de las pruebas de Fischer y t de Student, demostraron que no existían diferencias significativas entre las precisiones alcanzadas por los analistas en diferentes días para una probabilidad de 0,05%, ya que el valor de Fcalculada fue menor que la Ftabulada, estos resultados permitieron establecer que las precisiones son similares. Al realizar la prueba de t de Student el valor calculado resultó menor que el tabulado, para una probabilidad de 0,05, lo cual demostró que no existían diferencias significativas entre las medias alcanzadas, con un nivel de significación de un 5 %.
Los valores de porcentaje de recobrado estuvieron dentro de los límites establecidos para los métodos cromatográficos (98-102 %) y los valores del coeficiente de variación para cada uno de los valores de concentración estudiados resultaron ser menor que el 2 %. En la influencia del factor concentración sobre la variabilidad de los resultados de la exactitud al aplicar la prueba de Cochran se obtuvo que las G calculadas fueron menores que la G tabulada para una probabilidad de 0,05, k= 3 y n= 3; por lo tanto, las varianzas de las concentraciones empleadas son equivalentes indicando que la concentración no influyó en la variabilidad de estos. Al realizar la prueba de significación entre la recuperación media y el 100,0 % de recuperación, el valor de t calculada resultó menor que la t tabulada.
Estudio de estabilidad
El conjunto de los resultados del estudio de estabilidad acelerada a 40 ºC y 75 % de humedad relativa, demostraron la estabilidad del producto. Se demostró la estabilidad térmica del producto ya que después de transcurridos 6 meses se mantuvo la concentración conforme con los límites establecidos en las especificaciones de calidad del producto terminado.
De los resultados mostrados en la tabla 5 se infiere que el producto mantiene los parámetros que determinan su calidad, tanto en su etapa inicial como transcurridos 3, 6, 9, 12, 18 y 24 meses. Además, no se observan cambios en los aspectos organolépticos, el pH y la valoración de la concentración del principio activo durante el tiempo de almacenamiento estudiado. Los resultados de la esterilidad realizado el inicio y al final de estudio demostraron la estabilidad microbiológica del producto terminado. A partir de estos resultados se propusieron 2 años de validez, como tiempo de expiración para la formulación. No existieron diferencias significativas (p<0,05) entre las medias obtenidas en el tiempo inicial y el tiempo final de estudio.
En conclusión, el método analítico validado por CLAR para el estudio de estabilidad de la solución oral de risperidona 1 mg/mL, resultó específico, lineal, exacto y preciso en el intervalo de concentraciones estudiadas.
Los resultados de los estudios de estabilidad acelerada y de vida de estante, reportados en la Regulación 16-2000 del CECMED para Buenas Prácticas de Fabricación de Productos Farmacéuticos, demostraron la estabilidad física, química y microbiológica del producto por espacio de 24 meses a temperatura ambiente, ya que en todos los casos los resultados se encontraron dentro de los límites establecidos en las especificaciones de calidad de la solución oral de risperidona 1 mg/mL.
REFERENCIAS BIBLIOGRÁFICAS
1. Goodman A, Gilman A. Las bases farmacológicas de la terapéutica. Tomo II. 3ra. ed. La Habana: Editorial Científico Técnica; 1994. p. 423-8.
2. PDR. Physician´s Desk Reference. 57 ed. New York: Inc at Montuale; 2003. p. 2843.
3. Dierksneier G. Métodos cromatograficos. La Habana: Ed. Científico-Técnica; 2005. p. 1-4, 256-412.
4. Validation of Analytical Procedures. Technical Requirements for the Registration of Pharmaceuticals for Human Use. Geneve: International Conference on Harmonization, ICH-Q2A; 1995.
5. Validation of Analytical Procedures. Technical Requirements For The Registration of Pharmaceuticals For Human Use. Geneve: International Conference on Harmonization, ICH-Q2A; 1995.
6. United States Pharmacopoeial Convention. USP 30. United States Pharmacopoeia 30 and National Formulary No 25. Rockville: Marck Printing; 2007.
7. Regulación No. 16-2000: Buenas Prácticas de Fabricación de Productos Farmacéuticos. La Habana: Centro Estatal para el Control de Medicamentos (CECMED); 2000.
Recibido: 8 de abril de 2010.
Aprobado: 17 de mayo de 2010.
M. C. Caridad Margarita García Peña. Centro de Investigación y Desarrollo de Medicamentos (CIDEM). Ave. 26 No. 1 605 entre Boyeros y Puentes Grandes. CP 10 600. Plaza de la Revolución, La Habana, Cuba. Correo electrónico: caridadgp@infomed.sld.cu


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}