










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Farmacia
versión impresa ISSN 0034-7515
Rev Cubana Farm vol.47 no.1 Ciudad de la Habana ene.-mar. 2013
PRODUCTOS NATURALES
Ensayo de disolución para las tabletas de Tilo ® elaboradas con extracto seco de Justicia pectoralis Jacq
Dissolution Assay for Tilo ® tablets prepared with dry extract of Justicia pectoralis Jacq
DrC. Jorge Enrique Rodríguez ChanfrauI; Tec. Jose Manuel Gil Apan II.
I Centro de Investigaciones y Desarrollo de Medicamentos (CIDEM). La Habana, Cuba.
II Ministerio de Ciencia Tecnología y Medio Ambiente (CITMA). La Habana, Cuba.
RESUMEN
Introducción: el ensayo de disolución es una técnica analítica de empleo común en un laboratorio farmacéutico. Un proceso tecnológico para la elaboración de tabletas fue desarrollado. El ingrediente farmacéutico activo usado fue Tilo ® extracto seco.
Objetivo: el objetivo de este trabajo fue desarrollar y validar un ensayo de disolución para evaluar la estabilidad y la calidad de dicho producto.
Método: se utilizaron muestras de un lote experimental, un lote placebo y lotes pilotos de tabletas de Tilo ® de 100 mg. Se evaluaron como medios de disolución agua destilada y solución de ácido clorhídrico 0,1 mol/L, realizándose perfiles de disolución a 50, 75 y 100 rpm, empleándose los dos tipos de aparatos establecidos en la literatura para este ensayo (cesta y paleta). El contenido de cumarina fue analizado por HPLC. El ensayo fue validado según la USP.
Resultados: los resultados mostraron que el agua destilada fue un medio de disolución adecuado, alcanzándose porcientos de disolución de la droga por encima del 85 % a los 30 minutos, no existiendo diferencias significativas entre los tipos de aparatos recomendados por la USP. Mientras que, los perfiles de disolución a diferentes tiempos y velocidades de agitación mostraron una liberación gradual del principio activo en el tiempo, donde a medida que se incrementa la velocidad de agitación, se incrementa el porcentaje de disolución de la droga en el medio. La validación del ensayo demostró que el mismo era específico y preciso.
Conclusiones: se estableció como ensayo de disolución las siguientes condiciones de trabajo: Aparato: paleta, 100 rpm; medio: agua destilada, 500 mL; tiempo: 60 minutos y Temperatura: 37 ± 0,5 ºC.
Palabras clave: ensayo de disolución, perfiles de disolución, Justicia Pectoralis, tabletas, validación, cumarina.
ABSTRACT
Introduction: dissolution testing is one of the most common analytical techniques performed in a pharmaceutical analytical laboratory. A technological process for the production of tablets was developed. The active pharmaceutical ingredient used was Tilo ® dry extract.
Objective: to develop and to validate the dissolution assay aimed at evaluating the stability study and the quality of this product.
Methods: some samples from the experimental batch, the placebo batch and the pilot batches were used in this study. Distilled water and 0,1 mol/L chlorhidric acid were evaluated as dissolution media. The dissolution profiles at 50, 75 and 100 rpm and two types of dissolution devices (basket and paddle) recommended for the USP were evaluated. Coumarin content was analyzed by HPLC method. The dissolution assay was validated according to the United States Pharmacopeia.
Results: the results showed that the distilled water was an appropriate dissolution medium, where percentages of released drug higher to 85 % in 30 minutes were obtained; there were no significant differences among the types of dissolution devices recommended by the USP. The dissolution profiles at different shaking times and speeds showed gradual release of the active principle. As the shaking speed increases, the percentage of the drug dissolution increases in the medium. The assay was considered specific and precise.
Conclusions: a type II (paddle) dissolution device, 500 ml of distilled water at 37 ± 0,5 ºC and 100 rpm, were established as parameters of the dissolution assay.
Key word: dissolution test, dissolution profiles, justicia Pectoralis, tablets, validation, coumarin.
INTRODUCCIÓN
El ensayo de disolución es una técnica analítica de empleo común en un laboratorio farmacéutico. Se emplea generalmente en formulaciones orales con el objetivo de evaluar «in vitro» el comportamiento del principio activo en su forma terminada. Este ensayo es parte de otras pruebas analíticas empleadas para evaluar las formas terminadas farmacéuticas durante su desarrollo, estabilidad y control de la calidad.1
El estudio de los productos naturales a partir de plantas ha sido motivo de numerosas investigaciones a lo largo de muchos años. Una de las plantas que ha sido estudiada en nuestro país, por sus propiedades farmacológicas ha sido Justicia pectoralis Jacq, especie perteneciente a la familia de las Acanthaceae, abundante en las zonas tropicales de América, incluida Cuba.2-7
Se ha desarrollado un proceso tecnológico para la obtención de extracto seco de calidad farmacéutica a partir de extractos de esta planta medicinal, donde la cumarina es uno de los componentes principales del polvo. 8, 9 Esta materia prima ha sido empleada en el desarrollo de una forma terminada líquida (Jarabe) 10 y una forma terminada sólida (tabletas).11
El objetivo de este trabajo fue desarrollar y validar un ensayo de disolución de las tabletas de 100 mg de Tilo ® para su empleo como parte de los parámetros a controlar durante los estudios de estabilidad y los controles de calidad de dicho medicamento.
MÉTODOS
Para el desarrollo de este estudio se utilizaron muestras del lote experimental L-2, un lote placebo y los lotes pilotos 04001, 04002 y 04003 de tabletas de Tilo ® de 100 mg, elaboradas según el proceso tecnológico desarrollado en el Centro de Investigaciones y Desarrollo de Medicamentos (CIDEM). 11 como ingrediente activo farmacéutico se empleo Tilo ® extracto seco, obtenido de acuerdo a lo descrito por Rodríguez y col., 9 el cual cumplía los parámetros de calidad establecidos. 12 Como estándar de referencia se utilizo cumarina (Aldrich Chemical Co.).
Método de análisis
Un método de análisis por Cromatografía Líquida de Alta Resolución (HPLC), 12 basado en una modificación realizada al método propuesto por Rodríguez y col para la determinación de extractos secos obtenidos a partir de extractos de Justicia pectoralis Jacq. 13 fue empleado. El mismo fue previamente validado según las exigencias internacionales. 14
Se emplearon para ello las siguientes condiciones experimentales: Precolumna Aluspher® 100 (RP-select B 5mm) con Columna Lichrospher® 100, RP 18 (25 cm de longitud y 4 mm de diámetro y 5 micras), utilizándose como fase móvil una mezcla de metanol agua (40:60) y detector UV a una longitud de onda de 274 nm. El flujo de la fase móvil fue de 1 mL/min y el volumen de inyección fue de 20 mL.
Estudios de disolución
Selección del medio de disolución
Partiendo de los conocimientos previos que se tenían del comportamiento del principio activo, se decidió evaluar como medios de disolución agua destilada y solución de ácido clorhídrico 0,1 mol/L. Se realizaron pruebas empleando los dos tipos de aparatos establecidos en la literatura para este ensayo (cesta y paleta) y se compararon estadísticamente los resultados alcanzados. Las condiciones experimentales fueron:
Aparato: cesta o paleta, 50 rpm
Medio: agua destilada o solución de ácido clorhídrico 0,1 mol/L, 500 mL
Tiempo: 30 minutos
Temperatura: 37 ± 0,5 ºC
Perfiles de disolución
Seleccionado el medio y el aparato que mejores resultados mostraban en las condiciones experimentales antes mencionadas, se procedió a realizar los perfiles de disolución a 50, 75 y 100 rpm, tomándose muestras a los 15, 30, 60 y 90 minutos. En cada tiempo fueron extraídos 10 mL de solución de cada vaso disolutor, los cuales se reponían con medio de disolución a 37 ± 0,5 ºC y se filtraron. Se tomaron 5 mL de la muestra filtrada y se trasvasaron a un matraz aforado de 10 mL, llevándose a volumen con fase móvil, realizándose la determinación del contenido de cumarina disuelto en el mismo por el método de HPLC establecido.
Validación del ensayo de disolución
A partir de los resultados alcanzados en el estudio de los perfiles de disolución, se establecieron las condiciones del ensayo de disolución y se procedió a realizar la validación del mismo según las exigencias establecidas internacionalmente. 15, 16
La especificidad del ensayo se realizo con el empleo de 6 tabletas placebos y según el procedimiento establecido para el ensayo de disolución. Por otro lado, para evaluar la linealidad se preparo una curva de calibración en un intervalo de concentración desde 0,65 hasta 1,42 mg de cumarina (60 a 130% de la cantidad declarada). La significación del intercepto fue determinada mediante la prueba de Student, así como la prueba de linealidad fue aplicada. 15, 16
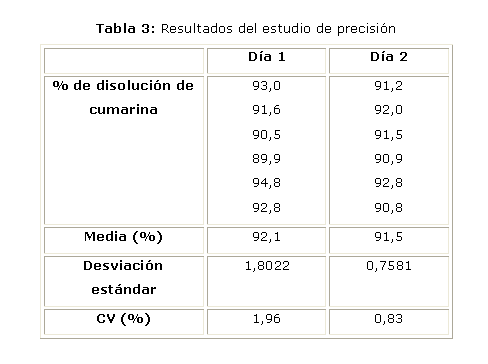
La precisión del ensayo fue evaluada empleando 6 tabletas del lote experimental en dos días diferentes.
Evaluación de los lotes pilotos
Finalmente los tres lotes pilotos fueron evaluados aplicando la metodología establecida para el ensayo de disolución desarrollado previamente.
Análisis estadístico
Los resultados obtenidos en los diferentes ensayos se procesaron estadísticamente utilizando los programas Statgraphics Plus (Versión 5.0) para Windows.
RESULTADOS
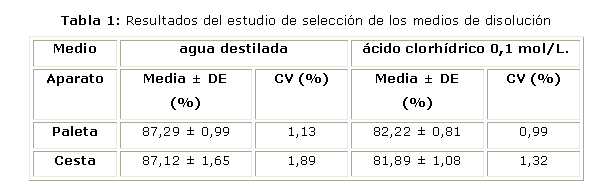
La tabla 1 muestra los resultados del estudio de selección del medio de disolución para los dos tipos de aparatos. El análisis estadístico de comparación de medias mostró que no existían diferencias significativas (p = 0,8297 cuando el medio es agua destilada y p = 0,5624 cuando el medio es ácido clorhídrico 0,1 mol/L) en los resultados obtenidos al emplearse paleta o cesta dentro de un mismo medio de disolución.
Por otro lado, al realizarse un análisis de varianza (ANOVA) para comparar ambos medios de disolución se comprobó que existen diferencias significativas (p = 0,000) entre ambos medios para un 95 % de confidencialidad. El procedimiento de Duncan de comparaciones múltiples identificó dos grupos homogéneos, que se corresponden cada uno con los medios de disolución estudiados, confirmando que los mismos son diferentemente significativos entre ellos.
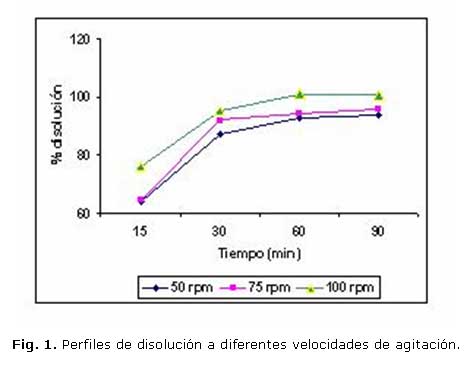
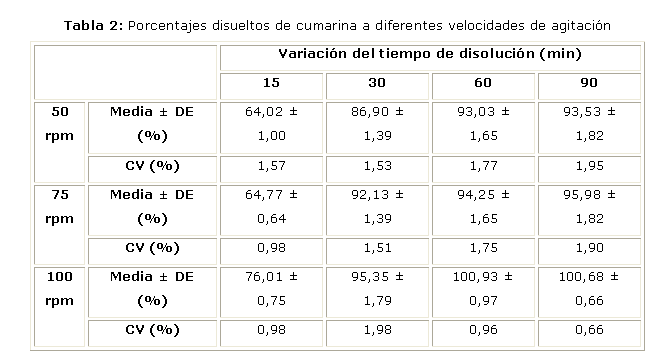
En la tabla 2 se muestran los resultados obtenidos a diferentes tiempos y distintas velocidades de agitación en el medio de disolución y el tipo de aparato seleccionado en el estudio anterior, mientras que la Figura 1 muestra los perfiles de disolución a diferentes velocidades de agitación.
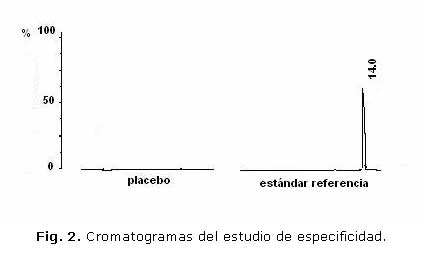
La prueba de especificidad del ensayo de disolución demostró que el análisis del placebo no muestra ningún pico en el cromatograma por lo que los excipientes presentes en la tableta no interfieren (Figura 2).
Al aplicar el método de los mínimos cuadrados a los resultados del estudio de linealidad, se obtuvo la ecuación de la recta que se expresa según y = 1,0048 X 0,0011, con un coeficiente de correlación lineal r = 0,9998 y un coeficiente de correlación ajustado r2 = 0, 9996 (criterios de aceptación r = 0,99 y r 2 = 0,98). 16 Al aplicar la prueba de Student para determinar la significación del intercepto, se obtuvo una t experimental = 0,091, p = 0,9334 menor que el valor tabulado (t tab (0,05, 13) = 2,16), por lo que se cumple la hipótesis nula de a = 0 para un nivel de confianza del 95 %. Mientras que los resultados para determinar la significación de la pendiente mostraron que t experimental = 0,441, p = 0,6889 menor que el valor tabulado (t tab (0,05, 13) = 2,16), cumpliéndose la hipótesis nula de b = 1 para un nivel de confianza del 95 %. Al calcular el coeficiente de variación de f se obtuvo un coeficiente de 1,06 % menor que el límite establecido del 5 %. 16
Los resultados del análisis de especificidad del ensayo se muestran en la tabla 3, observándose que en ambos casos los valores del coeficiente de variación son menores al 2 %, lo que es adecuado para este tipo de análisis. Al compararse mediante el test de Student las medias de cada día se comprueba que el valor experimental (texp = 0,004) es menor que el valor tabulado (t tab (0,05, 10) = 2,23).
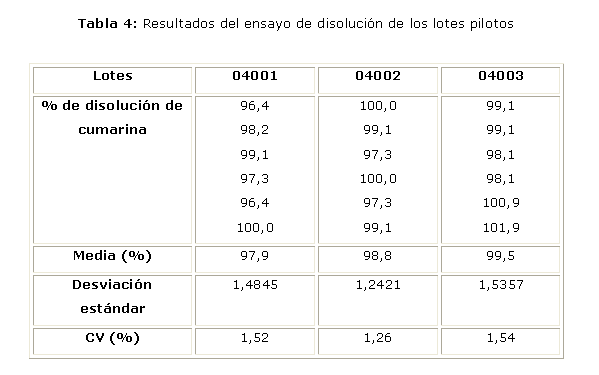
La tabla 4 muestra los resultados del ensayo de disolución de los lotes elaborados a escala piloto. En todos los casos los valores del porciento de disolución son superiores al 90 % y los valores de los coeficientes de variación son menores al 2 %.
DISCUSIÓN
El ensayo de disolución permite conocer la cantidad real de droga que se libera en un medio, por lo que es un ensayo que debe establecerse durante el proceso de desarrollo de las formas sólidas y mantenerse como parte del control de la calidad y estudio de estabilidad del medicamento. 17
Durante el desarrollo de una metodología para ensayo de disolución, el medio de disolución, el aparato y la velocidad de agitación deben ser evaluadas como parámetros necesarios para garantizar un ensayo que cumpla los requisitos establecidos para el buen funcionamiento del mismo. 1, 18
El medio de disolución es uno de los aspectos más importante, pues el mismo debe garantizar la solubilidad y recuperación de droga. Por otro lado, la velocidad de agitación también tiene un rol importante pues la misma debe garantizar un flujo adecuado que permita que la disolución sea gradual y no demasiado rápida. Se plantea que un perfil de disolución que presente los tres primeros puntos por debajo del 85 % de droga liberada es el ideal. 18
Los resultados de este trabajo mostraron que los mejores resultados de disolución se obtuvieron cuando el medio de disolución empleado fue agua destilada, alcanzándose porcientos de disolución de la droga por encima del 85 % a los 30 minutos, no encontrándose diferencias significativas entre los tipos de aparatos utilizados, por lo que se decidió establecer la paleta como aparato de disolución en el ensayo.
Al realizar los perfiles de disolución a diferentes tiempos y velocidades de agitación se comprobó que se logra una liberación gradual del principio activo en el tiempo, estando por encima del 85 % a partir de los 30 minutos. Se comprueba además que, a medida que se incrementa la velocidad de agitación, se incrementa el porcentaje de disolución de la droga en el medio.
Partiendo de estos resultados se estableció como ensayo de disolución las siguientes condiciones de trabajo: Aparato: paleta, 100 rpm; medio: agua destilada, 500 mL; tiempo: 60 minutos y Temperatura: 37 ± 0,5 ºC.
La USP 31 15 establece como parte del proceso de validación para el ensayo de disolución (categoría III) el estudio de la precisión como parámetro necesario a evaluar, mientras que los restantes parámetros se realizaran en dependencia de la naturaleza específica del ensayo.
Los resultados alcanzados en este estudio demostraron la especificidad del ensayo, al no detectarse que ninguno de los componentes de la formulación afectaba la cuantificación de la cumarina. Por otro lado, la curva de calibración se comportó de forma lineal en el rango de concentraciones evaluada, cumplimentando la ley de Lambert-Beer, aspecto comprobado por el alto valor del coeficiente de correlación alcanzado. Los resultados de la precisión demostraron que no existían diferencias significativas entre las medias.
Por todo lo anterior podemos afirmar que el ensayo propuesto es específico y preciso, cumplimentando las exigencias que para este tipo de análisis están establecidos a nivel internacional. 15, 16
La evaluación con la metodología establecida de los lotes pilotos demostró la factibilidad del ensayo para la determinación de la disolución en tabletas de Tilo ® 100 mg.
El ensayo de disolución para las tabletas de Tilo ® 100 mg, desarrollado resultó ser fiable teniendo en cuenta que el mismo cumple con los criterios establecidos en la literatura, por lo que se estableció como método de ensayo para los estudios de estabilidad y el control de la calidad de este medicamento.
REFERENCIAS BIBLIOGRÁFICAS
1. Chow Chan C; Rebelo-Cameirao A; Lee Y. C. Dissolution method validation. En: Chow Chan C; Lam H; Lee Y. C; Ming Zhang X. Analytical Method Validation and Instrument Performance Verification. Ed. John Wiley & Sons, Inc. New Jersey. 2004. p. 51 66.
2. Rivero R; Rodríguez Chanfrau J. E. Justicia pectoralis Jacq. Algunos aspectos sobre la composición química, farmacológica y toxicológica. Revista Mexicana de Ciencias farmacéutica. 2000; 30 (2): 15-19.
3. Roig, J. T. «Plantas Medicinales Aromáticas o Venenosas de Cuba». Ed. Científica y Técnica, La Habana, 1974. pp. 743.
4. Fernández L; Pérez H; Más R. Evaluación preliminar de los efectos neurofarmacológicos de la Justicia pectoralis Jacq. Revista Cubana Farmacia, 1989. 23 (1-2): 161-166.
5. Fernández, L; Pérez Saad H; Mas R; Rodríguez L; Galan L; Biscay R. Efecto de la Justicia pectoralis sobre la conducta exploratoria en los ratones. In: A. Álvarez y M. Valdés (eds), Estudios Avanzados de Neurociencias, Editorial del Centro Nacional de Investigaciones Científicas (CNIC), C. Habana, 1987. pp. 257-264.
6. Fernández L, Menéndez R, Fernández J; Más RM «Justicia pectoralis: Efectos sobre una tarea de evitación pasiva de una sola prueba en ratones». Rev. CNIC, Ciencias Biológicas. 1991. 22: 1-2.
7. Rodríguez EC, Virvés AT, Alemán SC L. "Estudio preliminar del efecto de la Justicia pectoralis sobre el EEG de adultos normales". Revista Cubana Farmacia. 1989. 23(3): 302-308.
8. Rodríguez Chanfrau J. E; López Hernández O. D; Núñez Y; Rodríguez C; Carrillo C; Gil Apan J. M; Echevarria I. Obtención de una Materia Prima de Calidad Farmacéutica a partir de Extractos de Justicia pectoralis Jacq., mediante Secado por Aspersión. Desarrollo Tecnológico a partir de Extracto Hidroalcohólico al 30%. Latin American Journal of Pharmacy. 2008. 27(3): 333-338
9. Rodríguez Chanfrau J, López Hernández OD, Núñez Y, Rodríguez C. Obtention of pharmaceutical quality raw material from aqueous extracts of Justicia pectoralis Jacq. by spray dried. Latin American Journal of Pharmacy. 2011. (en prensa)
10. de la Paz Martín-Viaña Nilia, Rodríguez Chanfrau Jorge, López Hernández Orestes D, González Sanabia María Lidia, Gil Apan José M, Fuste Moreno Viviana M et al . Desarrollo tecnológico de un medicamento sedante de origen natural a partir de Justicia pectoralis Jacq. Rev Cubana Plant Med [revista en la Internet]. 2011 Sep [citado 2012 Feb 19] ; 16(3): 227-235. Disponible en: http://scielo.sld.cu/scielo.php?script=sci_arttext&pid=S1028 -47962011000300002&lng=es.
11. Iraizos Barrios A. Tilo ® tabletas 100 mg. Informe final del diseño de una formulación. Informe técnico DTC/28. CIDEM. 2004
12. Rodríguez Chanfrau J. E; Fuste Moreno V. TILO EXTRACTO SECO MP. Técnica Nº PA 09005. CIDEM. 2009.
13. Rodríguez Chanfrau Jorge E., López Hernández Orestes D., Gil Apan José M.. Método para la cuantificación de cumarina en extracto seco a partir de extractos de Justicia pectoralis Jacq. Rev Cubana Plant Med [revista en la Internet]. 2008 Sep [citado 2012 Feb 19] ; 13(3): . Disponible en: http://scielo.sld.cu/scielo.php?script=sci_arttext&pid=S1028 -47962008000300004&lng=es.
14. Rodríguez Chanfrau JE, Fuste Moreno V. Validación analítica del método de análisis para la cuantificación de cumarinas totales en el medicamento tilo tabletas. Informe técnico VLM/09/11. CIDEM. 2011
15. United States Pharmacopoeia 31. US Pharmacopoeia Convention, Inc. Washington DC. 2008. p. 752 757.
16. ICH. Validation of Analytical Procedures: Text and Methodology. ICH Topic Q 2 (R1). CPMP/ICH/381/95. European Medicines Agency. London. 1995
17. ICH. A Critical View from the Generic Pharmaceutical Industry. Pharmaceutical Development. ICH Q8. Regulatory Requirements Directed by the New Note for Guidance (EMEA/CHMP/167068/2004) in Comparison to the Previous Guideline (CPMP/QWP/155/96). European Medicines Agency. London. 2004
18. Gray V; Zheng J; Sesi N. In vitro dissolution testing and method development. En: Zheng J. Formulation and Analytical Development for Low-dose Oral Drug Products. Ed. John Wiley & Sons, Inc. New Jersey. 2009. p. 265- 281
Recibido: 20 de agosto de 2012.
Aprobado: 21 de septiembre de 2012.
Jorge E. Rodríguez Chanfrau.
Centro de Investigaciones y Desarrollo de Medicamentos. Ave 26 # 1605. Nuevo Vedado. Ciudad de la Habana. CP 10600. Cuba. EMAIL: jorge.rodriguez@infomed.sld.cu

{kind=link}
{kind=link}
{kind=link}