










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Farmacia
versión impresa ISSN 0034-7515versión On-line ISSN 1561-2988
Rev Cubana Farm vol.48 no.3 Ciudad de la Habana jul.-set. 2014
ARTÍCULO ORIGINAL
Determinación de clorhidrato de nafazolina por cromatografía líquida de alta resolución en una solución oftálmica
Naphazoline hydrochloride assay by high performance liquid chromatography in an ophthalmic solution
MSc. Ernesto Benítez Hechavarría, MSc . Emilia Carolina Vargas Copland, MSc. German Eduardo Colindres Galindo, Lic. Klelia Felissia Enamorado Colindres
Laboratorios Andifar. Tegucigalpa, Honduras.
RESUMEN
Introducción: la Farmacopea de los Estados Unidos orienta cómo valorar clorhidrato de nafazolina en una solución oftálmica, pero indica el uso de una columna con grupos nitrilos unidos a sílice porosa, de uso poco frecuente.
Objetivo: desarrollar y validar un método alternativo por cromatografía líquida de alta resolución (CLAR) para la cuantificación de clorhidrato de nafazolina en una solución oftálmica.
Métodos: el método desarrollado fue de separación isocrática con columna Zorbax SB-C18 (4,6 x 150 mm, 5 µm) y detección ultravioleta a 225 nm. Como fase móvil se empleó solución amortiguadora y acetonitrilo (proporción 85:15, v/v) y la solución amortiguadora fue de KH2PO4 (22 mM) y trietilamina (30 mM), ajustada a pH 3 con ácido fosfórico concentrado. La validación del método se realizó siguiendo las indicaciones de la Guía Q2(R1) de la Conferencia Internacional sobre la Armonización. Se evaluaron los parámetros siguientes: especificidad, precisión, exactitud, linealidad y rango. También se determinó la incertidumbre del método.
Resultados: en la especificidad, el placebo no tuvo señal en la zona de clorhidrato de nafazolina; los coeficientes de variación obtenidos para la precisión intermedia resultaron inferiores a 1,5 %; en la exactitud el recobrado fue de 101,52 % y en la linealidad se demostró la ausencia de curvatura en el intervalo 50 a 150 %. La incertidumbre expandida calculada fue un 3 % de la cantidad declarada.
Conclusiones: todos los parámetros de validación evaluados se encuentran dentro los límites de aceptación establecidos, por lo que el método es adecuado para los fines propuestos.
Palabras clave: clorhidrato de nafazolina, cromatografía líquida de alta resolución, CLAR, validación, incertidumbre.
ABSTRACT
Introduction: the United States Pharmacopeia specifies how to titer naphazoline hydrochloride in an ophthalmic solution, but suggests the use of a column with nitrile groups attached to porous silica which is barely used.
Objective: to develop and to validate an alternative method by high resolution liquid chromatography for the quantification of naphazoline hydrochloride in an ophthalmic solution.
Methods: the developed method was isocratic separation with a Zorbax SB-C18 column (4.6 x 150 mm, 5 µm) and ultraviolet detection set at 225 nm. The mobile phase was buffer and acetonitrile (85:15 ratio, v/v) and the buffer was KH2PO4 (22 mM) and triethylamine (30 mM), adjusted to pH 3 with concentrated phosphoric acid. The validation method was performed pursuant to the Guide Q2(R1) of the International Conference on Harmonization and the following parameters were evaluated: specificity, precision, accuracy, linearity and range. The method uncertainty was also estimated.
Results: regarding the specificity, the placebo did not show any signal in the naphazoline hydrochloride zone; the relative standard deviation indexes for intermediate precision were less than 1.5 %; as to accuracy, the recovery was 101.52 % and the linearity showed absence of curvature in the 50 to 150 % range. The estimated expanded uncertainty reached 3 % of the stated amount.
Conclusions: all the validation parameters under evaluation were within the set allowable limits, thus this method is suitable for the intended purposes.
Keywords: naphazoline hydrochloride, High Performance Liquid Chromatography, HPLC, validation, uncertainty.
INTRODUCCIÓN
El clorhidrato de nafazolina (monoclorhidrato de 2-(1-naftilmetil)-2-imidazolina) es un potente agente simpaticomimético alfa-adrenérgico. Es un vasoconstrictor con una acción rápida y prolongada en la reducción de la hinchazón y congestión cuando se aplica a las membranas de la mucosa, por ello se utiliza para la liberación sintomática de la rinitis y sinusitis y en la descongestión de la conjuntiva ocular.1
En la Farmacopea Británica2 se describe un método para la valoración de clorhidrato de nafazolina (la sustancia activa) por cromatografía líquida de alta resolución (CLAR) pero no existe una monografía que indique el modo de valorar el clorhidrato de nafazolina en una solución oftálmica. En la Farmacopea de los Estados Unidos3 (USP, para sus siglas en inglés) existe una monografía que orienta la manera de valorar esta sustancia en una solución oftálmica, pero indican el uso de una columna L10 (grupos nitrilos unidos a sílice porosa) que no se posee en nuestro laboratorio.
El objetivo del presente trabajo fue desarrollar y validar un método alternativo para la cuantificación de clorhidrato de nafazolina en una solución oftálmica, con el propósito de ser utilizado para el control de calidad de esta solución.
MÉTODOS
QUÍMICOS Y REACTIVOS
Como sustancias químicas de referencia primaria y secundaria de clorhidrato de nafazolina se utilizaron: sustancia de referencia USP (lote M0K405) y sustancia de referencia del Laboratorio Andifar (lote A004), respectivamente. Las soluciones oftálmicas de clorhidrato de nafazolina y su placebo, se elaboraron en el Laboratorio Andifar. Los reactivos: fosfato monobásico de potasio, ácido fosfórico, ácido clorhídrico, hidróxido de sodio y acetonitrilo (grado CLAR), se adquirieron de Merck (Honduras). El etanol y la trietilamina se adquirieron de J. T. Baker (Honduras).
INSTRUMENTACIÓN Y CONDICIONES CLAR
En el ensayo se empleó un sistema cromatográfico (Perkin Elmer, series 200) con detector UV-visible ajustado a 225 nm. La separación fue isocrática con columna Zorbax SB-C18 (4,6 x 150 mm, 5 µm de tamaño de partícula). Se preparó una solución amortiguadora de KH 2PO4 (22 mM) y trietilamina (30 mM), ajustada a pH 3 con ácido fosfórico concentrado. Como fase móvil se empleó esta solución amortiguadora y acetonitrilo (proporción 85:15, v/v). La velocidad de flujo fue de 1 mL/min y se inyectaron 20 µL de muestra al sistema CLAR. La sustancia química de referencia y la muestra se prepararon a una concentración de clorhidrato de nafazolina de 40 µg/mL y fueron filtradas a través de filtros de 0,45 µm antes de la inyección.
VALIDACIÓN DEL MÉTODO
La validación se realizó siguiendo las indicaciones de la Guía Q2 (R1) de la Conferencia Internacional sobre la Armonización (ICH, para sus siglas en inglés).4 Según esta guía el método clasifica como ensayo cuantitativo de una sustancia activa en un producto terminado al cual se le deben evaluar las siguientes características de validación: especificidad, precisión (repetibilidad y precisión intermedia), exactitud, linealidad y rango.
Especificidad: se analizó el placebo y el placebo sometido a condiciones de estrés: hidrólisis básica (NaOH 0,1 N), hidrólisis ácida (HCl 0,1 N) y oxidación (H2O2 3 %), la relación placebo y solución de estrés fue de 1:1; todas se sometieron a 100° C durante 20 min, con reflujo. También se analizó la sustancia química de referencia secundaria sometida a hidrólisis básica a 100 °C, con reflujo, durante 5 min (se disolvieron 100 mg en 10 mL de etanol y luego se llevó a 100 mL con NaOH 0,1N).
Criterio de aceptación (CA): la señal, en los cromatogramas del placebo y placebos sometidos a estrés, en la zona correspondiente al clorhidrato de nafazolina debió ser menor a 0,5 % de la señal de la sustancia química de referencia y además que los productos de degradación de la hidrólisis básica de la sustancia química de referencia secundaria, no debieron interferir con la señal de clorhidrato de nafazolina.4
Precisión: incluyó la evaluación de la repetibilidad y la precisión intermedia.
- Repetibilidad: se realizó seis veces el método completo a una misma muestra, preparada al 100 % de la cantidad declarada.4 Se determinó el intervalo de confianza:
También se determinó el coeficiente de variación (CV).4,5
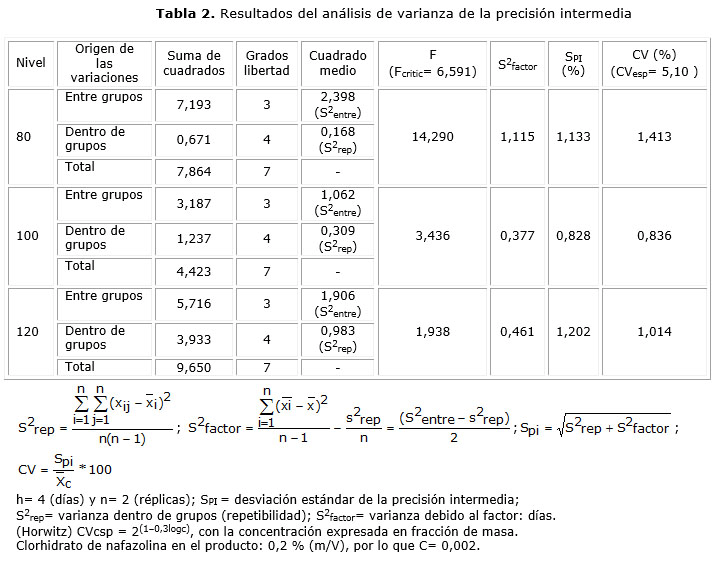
- Precisión intermedia: se analizaron muestras de soluciones oftálmicas al 80, 100 y 120 % de la cantidad declarada de clorhidrato de nafazolina. Las muestras fueron analizadas por duplicado, por dos analistas, en sistemas cromatográficos diferentes y en 4 días distintos (en un período de 2 meses que permitió abarcar las variaciones de reactivos, instrumentales y de condiciones climatológicas).4 Se realizaron las pruebas F y t de Student para determinar que no existieron diferencias significativas entre las variancias y los promedios obtenidos por los analistas, respectivamente.5 Se realizó un análisis de varianza de un factor (factor día) para las tres concentraciones. Se calculó el coeficiente de variación esperado (CVesp) utilizando la ecuación de Horwitz.6 Se determinaron los intervalos de confianza (IC) y los coeficientes de variación (CV).4,7 El CA para la repetibilidad fue que el CV debió ser inferior a 2,74 % (intervalo de aceptación 95-105 % y dos réplicas).7 El CA para la precisión intermedia fue que el CV obtenido para cada una de las concentraciones debió encontrarse por debajo o en el intervalo de 0,2 a 1 veces el CVesp.8
- Exactitud: se analizaron muestras de soluciones oftálmicas al 80, 100 y 120 % de cantidad declarada de clorhidrato de nafazolina (3 réplicas por concentración).4 Se determinó el recobrado y el intervalo de confianza del recobrado. Como CA se utilizó que la recuperación debió encontrarse entre 97 y 103 %.7
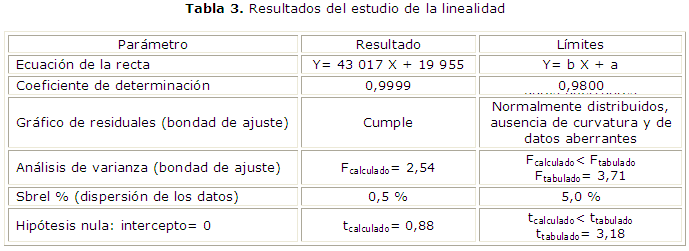
Linealidad: se analizaron cinco concentraciones (cada una por triplicado) obtenidas a partir de una solución madre de la sustancia química de referencia.4 Las concentraciones inyectadas fueron: 50, 80, 100, 120 y 150 %, donde el 100 % correspondió a 40 µg/mL de la sustancia química de referencia. Se determinaron: la ecuación de la recta y el coeficiente de determinación.4 Para comprobar la bondad de ajuste al modelo lineal se graficaron los residuales y además se comparó la varianza de la falta de ajuste contra la varianza del error experimental puro a través de un análisis de varianza, previamente se demostró homogeneidad de varianzas por Cochran (CA: los residuales debieron de encontrarse normalmente distribuidos, con ausencia de curvatura y de datos aberrantes; en el análisis de varianza el F calculado debió ser menor que el F tabulado, 3,71).8 La dispersión de los datos alrededor de la línea de regresión se determinó a través de desviación estándar relativa de la pendiente, Sbrel(%) (CA: el valor de Sbrel(%) debió ser menor que 5,0 %).8 Por último, para determinar si el intercepto era significativamente distinto de cero, se realizó la prueba t de Student (criterio de aceptación: el t calculado debió ser menor que el tabulado, 3,18).5,8
Rango: se estableció desde un 80 % hasta un 120 % de la concentración de ensayo: 40 µg/mL.4
Todos los datos obtenidos fueron procesados por el programa Microsoft Excel del paquete de programas Office y por el programa SPSS 15.0 para Windows.
DETERMINACIÓN DE LA INCERTIDUMBRE DEL MÉTODO
Se preparó una muestra control de clorhidrato de nafazolina, solución oftálmica, preparada al 100 % de la cantidad declarada. La incertidumbre del método se obtuvo combinando la incertidumbre asociada al recobrado y a la precisión intermedia.9-11 Para obtener la incertidumbre asociada al recobrado se analizó esta muestra control 10 veces, se calculó el recobrado, la desviación estándar de las 10 determinaciones y la incertidumbre asociada a la preparación de esta muestra control.10,11 La incertidumbre asociada a la precisión intermedia fue la desviación estándar obtenida luego de realizar varias determinaciones de esta muestra control en un período de 6 meses. 9,10
RESULTADOS
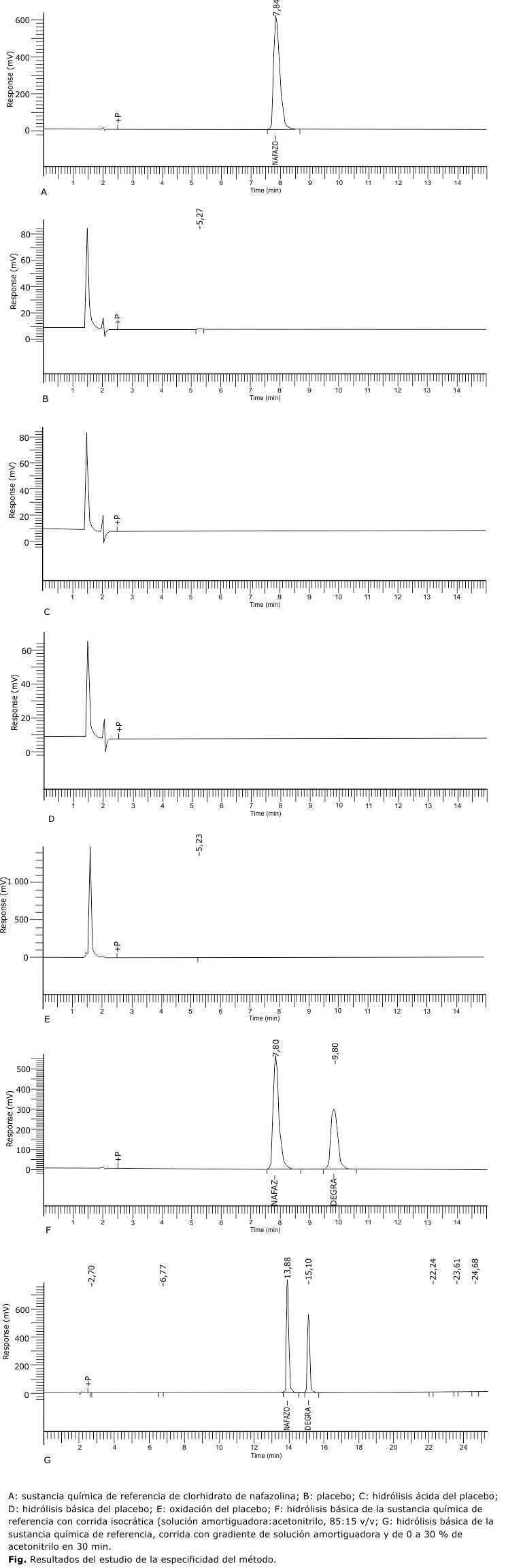
Los resultados del estudio de la especificidad se muestran en la figura. No se observó señal en la zona correspondiente al clorhidrato de nafazolina, ni en el cromatograma del placebo (B), ni en los cromatogramas del placebo sometido a estrés (C, D, E). Cuando se realizó la hidrólisis básica de la sustancia química de referencia secundaria de clorhidrato de nafazolina (F), se obtuvo un producto de degradación con tiempo de retención (TR) de 9,80 min, bien resuelto del pico de clorhidrato de nafazolina (TR= 7,80 min). Para determinar si existía algún otro producto de degradación básica que estuviera coeluyendo con el clorhidrato de nafazolina en el TR= 7,80 del cromatograma F, la misma muestra se corrió utilizando un gradiente (G) en lugar de una corrida isocrática. En el cromatograma G se obtuvieron los mismos picos obtenidos en F, pero con mayor número de platos teóricos.
En el estudio de la repetibilidad del método se obtuvo: promedio= 100,34 %; desviación estándar= 0,56 %; CV= 0,56 % y un intervalo de confianza de ± 0,59 %. Respecto a la precisión intermedia, los valores de F obtenidos en la prueba F fueron 2,79; 3,62 y 1,70 para las concentraciones de 80, 100 y 120 %, respectivamente, inferiores al tabulado: 9,28. En la prueba t de Student, los valores de t obtenidos fueron de 0,24 (80 %); 0,13 (100 %) y 1,70 (120 %), también inferiores al tabulado: 2,45. Los resultados obtenidos por cada analista se muestran en la tabla 1 y los resultados del análisis de varianza en la tabla 2.
En cuanto a la exactitud, la guía de la ICH para la validación de métodos analíticos4 especifica que esta debe reportarse como el % de recobrado (valor obtenido: 101,52 %) en conjunto con los intervalos de confianza (valores obtenidos: Media ± 0,53 %).
La tabla 3 muestra los resultados del estudio de la linealidad.
Los resultados de la cuantificación de la incertidumbre a partir de los datos obtenidos en los ensayos de la recuperación y de la precisión intermedia se muestran en la tabla 4.
DISCUSIÓN
En cuanto a la especificidad y como se evidencia en los resultados obtenidos, los componentes del placebo no interfieren en la determinación de clorhidrato de nafazolina. Respecto a la sustancia activa, se reporta que es estable en soluciones a pH ácidos y neutros y bajo condiciones de alta temperatura, expuesta a la luz y en soluciones saturadas con oxígeno. Sin embargo el clorhidrato de nafazolina muestra una degradación severa en soluciones alcalinas, estas soluciones se tornan oscuras en corto tiempo y como principal producto de degradación se obtiene el ácido naftil acético.1
En la hidrólisis básica de la sustancia química de referencia secundaria (cromatogramas G y F, figura) se obtuvo, además del pico de clorhidrato nafazolina, un pico que se supone corresponda a ácido naftil acético o a algún producto intermediario de esta degradación. Cuando se realizó el gradiente, los picos se hicieron más altos y estrechos y no apareció un pico adicional, lo cual indica que el pico de clorhidrato de nafazolina pudiera estar puro y que el producto de la degradación básica no interfiere en la determinación de este activo.
La guía ICH Q2 (R1) establece cómo debe ser evaluada la repetibilidad del método; sin embargo respecto a la precisión intermedia, no establece una pauta concreta. La repetibilidad del método cumple con el límite establecido; el CV fue inferior a 2,74 %, que es el límite cuando se realizan dos réplicas; y el intervalo de aceptación es de 95 a 105 % de la cantidad declarada,7 este es el intervalo establecido por control de calidad para la liberación de los lotes. En el caso de la precisión intermedia muchos autores prefieren determinar el CV global, es decir, considerando cada resultado de manera independiente.12,13 Otros autores prefieren separar la contribución de uno o más factores a la varianza global de la precisión intermedia.14,15
En la tabla 2 se observa que para las tres concentraciones se cumple con el límite establecido, es decir los CV de la precisión intermedia (1,41; 0,84 y 1,01 %) se encontraron por debajo o en el intervalo de 1,02 a 5,10 %. En el análisis de varianza (tabla 2), para la concentración de 80 %, el valor F calculado fue superior al crítico, lo cual indica que los promedios obtenidos en los diferentes días son distintos; en las concentraciones de 100 y 120 % el F calculado fue inferior al crítico, por lo que los promedios obtenidos en los diferentes días son iguales y la variabilidad total es explicada por la variabilidad dentro de los grupos, es decir, por la variabilidad de la repetibilidad. Respecto a los analistas, no existen diferencias significativas entre las varianzas obtenidas por ellos en ninguna concentración, los F calculados fueron inferiores al tabulado; tampoco existen diferencias en los promedios obtenidos donde el valor de t calculado fue inferior al tabulado en todos los casos.
Respecto a la exactitud, se obtuvo una recuperación cercana al 100 %. La recuperación obtenida se encuentra dentro de los valores aceptables para los formulados farmacéuticos (97-103).7
En cuanto a la linealidad, el gráfico de los residuales indica que estos se distribuyeron normalmente, sin datos aberrantes o tendencias hacia un mismo signo de los datos cercanos, también demuestra que la varianza es contaste en todo el intervalo de X, lo cual fue confirmado con la prueba de Cochran. Al ser la varianza independiente de la concentración, no hubo necesidad de realizar una regresión ponderada. En el análisis de varianza, el valor de F calculado fue menor que el tabulado, esto indica que la varianza de falta de ajuste no es estadísticamente mayor que la del error puro; esto quiere decir que no existe un error debido a la falta de ajuste que no pueda ser atribuido al error propiamente aleatorio, por lo que el modelo lineal se ajusta a los datos experimentales. Una vez demostrada la ausencia de curvatura, se evalúa la dispersión de los datos alrededor de la curva de calibración a través de la Sbrel (%), la cual fue muy inferior al límite recomendado de 5 %.8 Finalmente se demuestra que el intercepto no fue distinto de cero utilizando la prueba t de Student.
La incertidumbre estándar combinada obtenida representa un 1,5 % de la concentración de la muestra. La precisión intermedia del método introduce tres veces más incertidumbre que el recobrado o trazabilidad a juzgar por los datos obtenidos de incertidumbre estándar relativa.
El propósito de este método es la cuantificación de clorhidrato de nafazolina en una solución oftálmica, este producto terminado se libera en producción si cumple con una especificación para la potencia de 95 a 105 % de la cantidad declarada (± 5 %). Todos los parámetros de validación evaluados se encontraron dentro los límites de aceptación establecido y la incertidumbre expandida calculada fue de 3 % de la cantidad declarada, por lo que el método es adecuado para los fines propuestos.9 Los lotes de producción serán rechazados si el resultado obtenido más/menos la incertidumbre del método se encuentra fuera de la especificación para la potencia.
REFERENCIAS BIBLIOGRÁFICAS
1. Wall GM. Naphazoline hydrochloride. En: Brittain HG, editor. Analytical profiles of drug substances and excipients. Vol 21. San Diego: Academic Press, Inc; 1992. p. 307-44.
2. British Pharmacopoeia [CD-ROM]. 7a ed. London: The Stationery Office; 2009.
3. United States Pharmacopoeial Convention. USP XXXV. United States Pharmacopoeia. 35 ed. Rockville: United Book Press; 2012. p. 4454-55.
4. International Conference of Harmonization (ICH) Q2(R1) Validation of Analytical Procedures: Text and Methodology. November 2005. p. 1-13. [cited 2013 Aug 16]. Available from: http://www.ich.org/products/guidelines/quality/article/quality-guidelines.html
5. Miller JC, Miller JN. Lyman R. Estadística para Química Analítica. 2da ed. Wilmington: Addison-Wesley Iberoamericana; 1993.
6. Horwitz W. Evaluation of analytical methods used for regulation of foods and drugs. Anal Chem. 1982 Jan;54(1):67-76.
7. Aguirre L, García FJ, García T, Illera M, Juncadilla M, Lizardo M, et al. Validación de Métodos de Análisis en Materias Primas y Especialidades Farmacéuticas. En: Pérez JA, Pujol M. Validación de Métodos Analíticos. Barcelona: AEFI; 2001. p. 45-133.
8. González AG, Herrador MA. A practical guide to analytical method validation, including measurement uncertainty and accuracy profiles. Trends in Analytical Chemistry. 2007;26(3):227-38.
9. Magnusson B, Näykki T, Hovind H, Krysell M. Handbook for calculation of measurement uncertainty in environmental laboratories. Oslo, Noruega: Nordic Innovation Stensberggata 25 NO-0170; 2012. NT techn report 537, edition 3.1. [cited 2013 Aug 16]. Available from: http://nordtest.info/index.php/technical-reports/category/environment.html
10. Barwick V, Ellison S. Protocol for uncertainly evaluation from validation data. United Kingdom: National Measurement Systems Valid Analytical Measurement (VAM) Programme; 2000. VAM Project 3.2.1, version 5.1. [cited 2013 Aug 16]. Available from: http://blpd.dss.go.th/knowledge_el/VAM_uncertainty-0452.pdf
11. Ellison S, Williams A. Quantifying Uncertainly in Analytical Measurement. Eurachem/CITAC guide. 3ra ed. 2012. [citado 16 ago 2013]. Disponible en: http://www.eurachem.org/index.php/publications/guides/quam
12. Jiménez NM, Calero JE, Padrón AS, Izquierdo JC. Método analítico por cromatografía de alta resolución para la determinación de carbamazepina en plasma humano. Rev Cubana Farm. 2007 [citado 30 jul 2013];41(1). Disponible en: http://scielo.sld.cu/scielo.php?pid=S0034-75152007000100007&script=sci_arttext
13. García CM, Pereda D, González A, Montes de Oca Y, Cañizares Y, León GM. Determinación de diclofenato de sodio por cromatografía líquida de alta resolución en un colirio al 0.1 %. Rev Cubana Farm. 2009 [citado 30 jul 2013];(3). Disponible en: http://scielo.sld.cu/scielo.php?pid=S0034-75152009000300006&script=sci_arttext
14. Boqué R, Maroto A, Riu J, Rius FX. Validation of analytical methods. Grasas y Aceites. 2002;53(1):128-43.
15. Thompson M, Ellison S, Wood R. Harmonized guidelines for single-laboratory validation of methods of analysis (IUPAC technical report). Pure Appl Chem. 2002;74(5):835-55.
Recibido: 10 de julio de 2014.
Aprobado: 15 de agosto de 2014.
Ernesto Benítez Hechavarría. Laboratorio Andifar. Col. Kennedy, Zona Industrial Jacaleapa, Carretera a UNITEC, Tegucigalpa, Honduras. Correo electrónico: ebhrria@yahoo.com


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}