










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista

Articulo
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Medicina
versión impresa ISSN 0034-7523versión On-line ISSN 1561-302X
Rev cubana med v.42 n.6 Ciudad de la Habana nov.-dic. 2003
Hospital Clinicoquirúrgico " Cmdte. Manuel Fajardo." Servicio de Neurología
Hospital Clinicoquirúrgico "Hermanos Ameijeiras." Servicio de Neurología
Variante asimétrica de la polineuropatía desmielinizante inflamatoria crónica. Presentación de 1 caso
Dr. Claudio Enrique Scherle Matamoros,1 Dr. Alejandro Negrin Expósito,1 Dr. Jesús Pérez Nellar2 y Dr. Carlos Maya Entenza3
Resumen
Se describió el cuadro clínico de un paciente con una variante asimétrica de polineuropatía desmielinizante inflamatoria crónica, con predominio distal en los miembros superiores, sin respuesta a varias modalidades de tratamiento inmunosupresor.
DeCS: POLINEUROPATIA/etiología; POLINEUROPATIA/terapia.
La polineuropatía desmielinizante inflamatoria crónica (PDIC) clásica se define como una afección neuropática simétrica de predominio motor con debilidad proximal y distal, asociada a pérdida de los reflejos osteotendinosos de curso progresivo o recurrente. Cursa con aumento del contenido de proteínas en el líquido cefalorraquídeo, así como evidencias de desmielinización en los estudios de neurografía y morfológicos.1
Para realizar el diagnóstico se emplean los criterios propuestos por la Academia Americana de Neurología,2 que si bien permiten hacer una selección homogénea de casos con fines investigativos, paralelamente no permiten "incluir" en la práctica clínica diaria a casos que tienen fuertes evidencias de una neuropatía desmielinizante adquirida con manifestaciones que difieren de la forma clásica de las PDIC.
La heterogeneidad clínica de la PDIC ha sido reconocida recientemente.3 El fenotipo clínico asimétrico mixto corresponde a una categoría diferente de la variante clásica de PDIC, denominado neuropatía sensorimotora desmielinizante adquirida multifocal, en la que el tratamiento con esteroides o inmunoglobulina parenteral resulta de elección.3,4
Describimos el cuadro clínico de un paciente con una variante asimétrica de PDIC con predominio distal en los miembros superiores, sin repuesta a varios esquemas de tratamiento inmunosupresor.
Caso clínico
Presentamos el caso de un varón, blanco, de 45 años de edad, con antecedentes de HTA leve tratada con clortalidona (25 mg/d) y propranolol (80 mg/d). Ocupación: cocinero. Sin otros datos de exposición a tóxicos o drogas relacionadas con enfermedades neuromusculares, ni antecedentes familiares de neuropatías hereditarias.
A finales del año 1997 comienza a sentir parestesias (sensación de hormigueo, entumecimiento y pinchazos) en el territorio del nervio cubital y mediano de la mano derecha, que en un período de 6 meses progresan hasta afectar la distribución del nervio mediano contralateral. Un año después le aparecen parestesias y debilidad en el pie derecho que evolutivamente le dificultan la marcha y el ascenso de escaleras.
Al examinarlo, en el mes de mayo del 2000, encontramos:
Marcha en stepage. Reflejos osteotendinosos globalmente hipoactivos.1 Debilidad (4 /5) de la flexión de los dedos de la mano derecha, así como debilidad (2/5) de la flexión dorsal y (4/5) de la extensión del pie derecho.
Hipoestesia superficial en el territorio de ambos nervios medianos (D>I) e hipoestesia superficial cubital derecha.
Hipopalestesia y disbatiestesia en los 3 primeros dedos de la mano derecha (fig.).
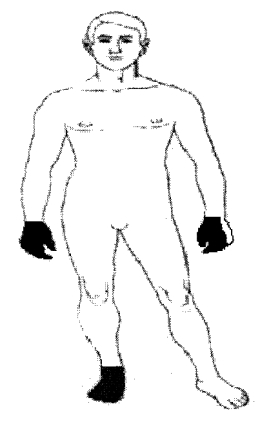
Figura. Patrón clínico de afectación .
Nervio cubital derecho palpable a nivel del codo y muñeca
En el estudio de neuroconducción por tramos, en el que se examinan los nervios medianos, cubitales, radiales, peroneos y tibiales, encontramos hallazgos compatibles con mononeuropatía múltiple predominantemente motora, con afectación axonal moderada y mielínica severa (tabla).
Tabla. Resumen de los datos obtenidos en el estudio de neurografía
| Nervios | Hallazgos |
| · Medianos (motor y sensitivo) | Daño axonal y mielínico severo |
| · Peroneo derecho | Daño axonal y mielínico severo |
| ·Tibial derecho | Daño mielínico severo |
| ·Tibial izquierdo | Daño axonal y mielínico severo |
En el mes de marzo, iniciamos el tratamiento con prednisona (100 mg/d), sin obtener respuesta después de 6 meses. Se añade entonces ciclofosfamida 90 mg/d durante 3 meses. En marzo del 2001 es ingresado y se le administra intacglobin (IgG) EV a 400 mg/kg en un período de 5 d. Nuevamente es reevaluado al mes del alta sin evidencias de mejoría clínica. Se decide administrar nuevamente la prednisona a razón de 1 mg\kg de peso y en octubre del mismo año se repite el ciclo de intacglobin (IgG) EV. En evaluaciones posteriores, los hallazgos clínicos y neurofisiológicos han evidenciado la progresión de su enfermedad.
Investigaciones complementarias
Hemoglobina: 146 g/L; hematócrito: 0,42; velocidad de sedimentación globular: 10 mm/h; conteo global de leucocitos: 9,0 x 10 9; glucemia: 5,2 mmol/L; creatinina:99 mmol/L; transaminasa glutámico-piruvica: 5 U/L; bilirrubina total: 3,8 y directa: 2,2; Proteínas totales y fraccionadas: proteínas 72, albúmina 44, globulina: 28; AgSHB : negativo; anti-HVC: negativo. Parcial de orina: sin alteración. VDRL: no reactivo. HIV: negativo. ANA: negativo, crioglobulinas: negativas, factor reumatoideo: negativo, ICC: 0,45 (valor de referencia: 0,01-0,042), CH 50 : 32 (valor de referencia 29±3), electroforesis de proteínas séricas: no mostró proteína M.
Estudio citoquímico del LCR: proteínas 0,65 g/L; glucosa: 3,6 mmol/L; células blancas: 0. No se determinaron anticuerpos anti GM1 por no contar con este recurso en el laboratorio de hospital.
Rayos X de tórax sin alteraciones pleuropulmonares, área cardíaca dentro de límites normales, baciloscopia Hansen: codificación 0. Electrocardiograma dentro de límites normales.
Discusión
El examen físico, la neurografía y el estudio histológico de nervio periférico confirman el compromiso multineurítico mixto, crónico, desmielinizante con predominio en el hemicuerpo derecho, lo que define el diagnóstico como una neuropatía desmielinizante multifocal sensitivo motora adquirida.
Entre las causas de mononeuritis múltiple que debemos excluir se encuentran las vasculitis. La falta de manifestaciones sistémicas asociadas y los resultados negativos de los estudios de laboratorio hacen poco probable este diagnóstico etiológico. Por otro lado, los hallazgos desmielinizantes encontrados contrastan con la degeneración axonal característica de las mononeuritis múltiples provocadas por vasculitis.5
Los signos de daño axonal en este paciente pueden estar en relación con el tiempo de evolución de la afección neuropática y ser secundarios al proceso de desmielinización.6 El deterioro crónico de la mielina conduce a disminución del número de neurofilamentos y de los niveles de fosforilación en el axón, con subsecuente disminución del diámetro y de su transporte.7
En 1982, Lewis y otros8 describieron 5 pacientes con una forma atípica de mononeuritis múltiple sensitivo motora, desmielinizante con predominio en los miembros superiores y de comienzo insidioso. La progresión en todos los casos fue lenta y los niveles de proteínas en el LCR eran normales o estaban ligeramente aumentados. En los estudios neurofisiológicos se hallaron signos de desmielinización y la presencia de bloqueos múltiples de la conducción motora en sitios que no son de atrapamiento. Dos de los sujetos presentados fueron tratados con prednisona exhibiendo mejoría posterior.
Casos similares a los de Lewis han sido publicados posteriormente9-12 y tanto la prednisona como la IgG EV se han utilizado en el tratamiento. Sin embargo, entre el 30 y 50 % de los casos no responden a la terapia con esteroide y la inmunoglobulina no resulta efectiva en un 30 %.10,11
Summary
The clinical picture of a patient with an asymmetric variant of chronic inflammatory demylienating polyneuropathy with distal predominance in the upper limbs and without response to various modalities of immunosupressive treatment, was described.
Referenias bibliográficas
- Barohn RJ, Kissel JT, Warmolts JR, Mendell JR. Chronic inflammatory demyelinating polyradiculoneuropathy: clinical characteristics and recommendation for diagnostic criteria. Arch Neurol 1989;46:878-84.
- Cornblath DR, Asbury AK, Albers JW, Feasby TE, Hahn AF, Mc Leod JG et al. Report from an Ad Hoc Subcommitee of The American Academy of Neurology AIDS Task Force. Research criteria for the diagnosis of chronic inflammatory demyelinating polyneuropathy (CIDP). Neurology 1991;41:617-8.
- Rotta FT, Sussman AT, Bradley WG, Ayyar DR, Sharma KR, Shebert RT. The spectrum chronic inflammatory demyelinating polyneuropathy. J Neurol Sciences 2000;173:129-39.
- Saperstein DS, Katz JS, Amato AA, Barohn RJ. Clinical spectrum of chronic acquired demyelinating polyneuropathies. Muscle Nerve 2001;24:311-24.
- Rosembaum R. Neuromuscular complications of connective tissue diseases. Muscle Nerve 2001;24: 154-69.
- Adams RD, Victor M, Ropper AH. Diseases of the peripheral nerves. Principles of Neurology. 6 ed. New York: McGraw-Hill; 1997. p. 1302-69.
- Martini R. The effect of myelinating schwann cells on axon. Muscle Nerve 2001;24:456-66.
- Lewis RA, Sumner AJ, Brown MJ, Asbury AK. Multifocal demyelinating neuropathy with persistent conduction block. Neurology 1982;32:958-64.
- Gibbels E, Behse F, Kentenich M, Haupt WF. Chronic multifocal neuropathy with persistent conduction block (Lewis-Sumner syndrome). Clin Neuropathol 1993;12:343-52.
- Oh SJ, Claussen GC, Kim DS. Motor and sensory demyelinating mononeuropathy multiplex (multifocal motor and sensory demyelinating neuropathy): a separate entity or a variant of chronic inflammatory demyelinating polyneuropathy?. J Peripheral Nervous System 1997;2:362-9.
- Saperstein DS, Amato AA, Wolfe GI, Katz JS, Nations SP, Jackson CE, Bryan WW, Burns DK, Barohn RJ. Multifocal acquired demyelinating sensory and motor neuropathy: the Lewis-Sumner syndrome. Muscle Nerve 1999;22:560-6.
- Van den Berg-Vos RM, Van den Berg LH, Franssen H, et al. Multifocal inflammatory demyelinating neuropathy: a distinct clinical entity?. Neurology 2000;54:26-32.
Recibido: 22 de mayo de 2003. Aprobado: 22 de abril de 2003.
Dr. Claudio Enrique Scherle Matamoros. Calle 24 No. 307 entre 19 y 21, El Vedado, Ciudad de La Habana, Cuba. Correo electrónico: csm@infomed.sld.cu
1 Especialista de I Grado en Neurología. Especialista de I Grado en Medicina General Integral. Hospital Clinicoquirúrgico " Cmdte. Manuel Fajardo."
2 Especialista de II Grado en Neurología. Hospital Clinicoquirúrgico "Hermanos Ameijeiras."
3 Especialista de I Grado en Neurología. Hospital Clinicoquirúrgico " Cmdte. Manuel Fajardo."

