










Meu SciELO
 Serviços customizados
Serviços customizadosServiços Personalizados
Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkRevista Cubana de Medicina
versão On-line ISSN 1561-302X
Rev cubana med v.49 n.1 Ciudad de la Habana jan.-mar. 2010
PRESENTACIÓN DE CASOS
Enfermedad de Creutzfeldt-Jakob en un paciente interpretado como encefalitis por herpes simple
Creutzfeldt-Jakob disease in a patient with possible herpes simplex encephalitis
Edmundo Rivero AriasI; David Lozano ValdésII; David Cubero RegoIII; Alfredo Sánchez ValdiviaIV
IEspecialista de II Grado en Neurología. Especialista de II Grado en Medicina Intensiva y Emergencias. Máster en Infectología. Profesor Auxiliar. Investigador Auxiliar. Hospital Clínico Quirúrgico "Hermanos Ameijeiras". La Habana, Cuba.
IIEspecialista de II Grado en Medicina Interna. Profesor Auxiliar. Hospital Clínico Quirúrgico "Hermanos Ameijeiras". La Habana, Cuba.
IIIEspecialista de II Grado en Anatomía Patológica. Asistente. Hospital Clínico Quirúrgico "Hermanos Ameijeiras". La Habana, Cuba.
IVEspecialista de I Grado en Medicina Intensiva. Hospital Clínico Quirúrgico "Hermanos Ameijeiras". La Habana, Cuba.
RESUMEN
Se presentó un paciente con una enfermedad de Creutzfeldt-Jakob, al cual se le realizó un diagnóstico presuntivo inicial de encefalitis por herpes simple, que comenzó con trastornos conductuales, agresividad, alucinaciones visuales y pérdida progresiva de la visión, empeoró de forma paulatina, aparecieron movimientos involuntarios inespecíficos y estado de postración, convulsiones tónico-clónicas del hemicuerpo izquierdo, paresia homolateral, poca respuesta al dolor con empobrecimiento de la mímica facial e imposibilidad para localizar el estímulo nociceptivo y discreta rigidez nucal. Se le realizaron estudios microbiológicos para pesquisaje virológico incluido herpes virus, bacterias y hongos, así como análisis citoquímico del líquido cefalorraquídeo y resultaron normales, en el electroencefalograma aparecieron alteraciones que señalaron actividad paroxística asimétrica. El paciente evolucionó tórpidamente y falleció, se presenta el resultado de los estudios anatomopatológicos de la microscopia óptica.
Palabras clave: Enfermedad de Creutzfeldt-Jakob, trastornos conductuales, estudios microbiológicos, convulsiones, electroencefalograma, rigidez nucal.
ABSTRACT
This is the case of a patient presenting with Creutzfeldt-Jakob disease with an initial presumptive diagnosis of herpes-simplex encephalitis started with behavioral disorders, aggression, visual hallucinations and a progressive loss of vision, worsening in a gradual way, non-specific unintentional movements and prostration status, left hemi-body tonic-clonic convulsions, homolateral paresis, a low response to pain with a poor facial expression and inability to locate the nociceptive stimulus and a light nuchal stiffness. Microbiologic studies were conducted for a virology screening including herpesvirus, bacteria and fungi, as well as, cytochemistry of cerebrospinal fluid with normal results, in electroencephalogram appeared alterations related to an asymmetric paroxysmal activity. Patient evolved with torpidity and died. Anatomic-pathologic studies result of optical microscopy is showed.
Key words: Creutzfeldt-Jakob disease, behavioral disorders, microbiologic studies, convulsions, electroencephalogram, nuchal stiffness.
Antecedentes históricos de las enfermedades producidas por priones
La enfermedad de Creutzfeldt-Jacob (ECJ) es un enfermedad degenerativa del sistema nervioso central, de curso fatal, que se presenta con deterioro cognitivo, alteraciones neuropsiquiátricas, mioclonias, postración, coma y muerte, se encuentran en el examen anatomopatológico cambios espongiformes con vacuolización intraneuronal, gliosis y ausencia de reacción inflamatoria en los tejidos afectados, debida en general a un trastorno a nivel del cromosoma 20 con una consiguiente distorsión en el plegamiento de una proteína, llamada priónica, se ha definido hasta ahora la variante esporádica, la familiar, la iatrogénica y la llamada nueva variante.1
En 1920, Jakob describe varios casos de una rara enfermedad neurológica crónica, cuya característica común era la demencia,2 muy parecidos a un caso al cual Creutzfeldt ya había hecho referencia un año antes.3 Fue el patólogo islandés Sigurdsson en el año 1954 el primero que empleó el termino infección lenta refiriéndose a una enfermedad que afectaba a las ovejas llamada scrapie;4 Gajdusek y Zigas en el año 1957 reportaron en la tribu Fore, de Nueva Guinea, un tipo de encefalopatía en la que el denominador común era el canibalismo ritual, el Kuru.5 Poco después un veterinario, Hadlow, encuentra puntos de coincidencias entre el Kuru y el scrapie.4
En los años 60 se logró inocular con el Kuru a animales de experimentación y la ECJ, logrando demostrar el carácter infeccioso de ambas entidades. En 1982 Prusiner planteó la teoría de las proteínas infecciosas o priones (proteinaceus infectious particle) como causa de esta enfermedad y tres años más tarde se descubrió que la proteína priónica es un componente normal de la célula codificada en el brazo corto del cromosoma 20.6
Ocurre la primera epizootia de enfermedad espongiforme bovina en el 1986 en el Reino Unido y diez años más tarde se reporta el primer caso de nueva variante del ECJ, también en la Gran Bretaña. Otras formas de enfermedad priónica han sido descritas, entre ellas, el síndrome de Gerstmann-Sträussler-Scheinker y el insomnio familiar fatal, que aunque más raras, conforman este temible grupo de enfermedades, para las cuales no existe cura hasta el momento y evolucionan inexorablemente a la muerte.7
En 1999 en Cuba se presentó un paciente con enfermedad de ECJ en su variante esporádica correspondiendo con la baja incidencia de esta enfermedad señalada por varios autores, es común el diagnóstico clínico equívoco al comienzo de los síntomas, por otra parte se señala su semejanza con otras entidades causantes de demencia.8
PRESENTACIÓN DEL CASO
Paciente masculino, de 74 años de edad, campesino, jubilado, de raza blanca, con antecedentes de buena salud anterior, sus familiares notan que desde hace aproximadamente dos meses antes de su ingreso comenzó con trastornos de conducta, agresividad, alucinaciones visuales y pérdida progresiva de la visión, fue ingresado con este cuadro clínico, empeorando de forma paulatina, con la aparición de movimientos involuntarios inespecíficos y estado de postración, fue interpretado como una encefalitis por herpes simple y se trasladó al Servicio de Cuidados Intensivos, donde se constataron convulsiones tónico-clónicas del hemicuerpo izquierdo, paresia homolateral, poca respuesta al dolor con empobrecimiento de la mímica facial e imposibilidad para localizar el estímulo nociceptivo, a lo que se le añade discreta rigidez nucal, se inicia tratamiento con aciclovir a dosis de 10 mg por kilogramo de peso.
El paciente evolucionó de forma tórpida, con una bronconeumonía bacteriana nosocomial a germen no identificado, presentó una insuficiencia respiratoria agravada por un tromboembolismo pulmonar y falleció por esta causa. El diagnóstico de enfermedad de ECJ fue hecho en vida por el método clínico, pero no se llegó a realizar biopsia cerebral por el estado crítico que presentaba el paciente, los hallazgos anatomopatológicos de microscopía óptica realizado luego de la necropsia donde se aplicaron varias tinciones a los tejidos obtenidos, se muestran en el presente trabajo.
Estudios realizados
Se realizó examen citoquímico del líquido cefalorraquídeo que resultó normal, con estudio bacteriológico, micológico, BAAR y pesquisaje virológico incluido Herpes virus que resultaron normales.
El electroencefalograma realizado resultó anormal, con asimetría interhemisférica, signos de irritación cortical focal intercrítica de intensidad moderada en la región anterolateral del hemisferio derecho y atenuación o ausencia unilateral del ritmo alfa sobre todo en ese hemisferio.
En la tomografía axial computarizada solo se encontró ligera atrofia cortical frontal sin otras alteraciones.
Los exámenes complementarios de laboratorio que se realizaron, que incluyeron hemograma con diferencial, glicemia, pruebas funcionales hepáticas, y estudio de la función renal, fueron normales, se evidenció solo un aumento de los leucocitos polimorfonucleares coincidiendo con infecciones intercurrentes.
Los cambios patológicos observados pueden verse en las figuras 1, 2 y 3.

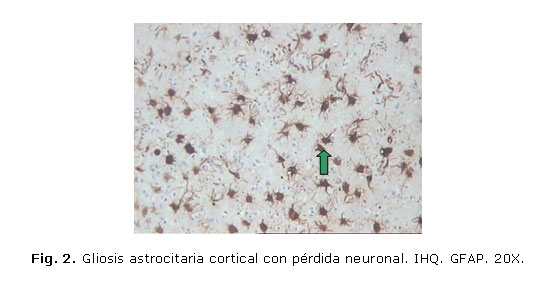
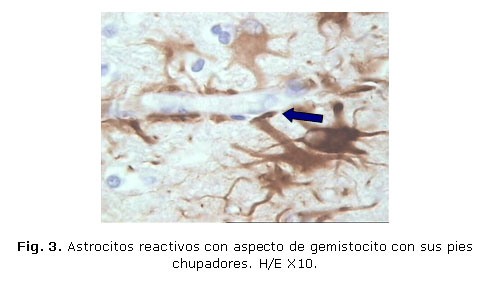
DISCUSIÓN
El diagnóstico de esta rara entidad se debe realizar siempre que existan criterios clínicos para plantearla; las posibilidades de realizar una biopsia del encéfalo u otras pruebas confirmatorias en el paciente vivo es de particular importancia debido a que entre otras cosas, descarta otras enfermedades tratables. Así, la integración de especialidades como Neurología, Neurocirugía y Anatomía Patológica, es de suma importancia para el diagnóstico de las enfermedades por priones en general.
Por otra parte, en lo relacionado con el aspecto de la manipulación del material de laboratorio el cual en su mayoría deberá ser desechado, existen ciertos equipos relacionados con el diagnóstico de laboratorio que son reutilizables, solo que se debe proceder a una desinfección particular de acuerdo al caso.
Los materiales infectados deben ser esterilizados en una solución de NaOH por 1 h a temperatura ambiente, se puede contar además con la esterilización con autoclave por aproximadamente 4,5 h a 121 °C, no es recomendable la esterilización con irradiación ultravioleta, alcohol, formalina o fenol.
El temor a contraer esta entidad a veces puede entorpecer el diagnóstico en la práctica clínica diaria, por lo que la observancia de estas medidas de desinfección, así como el cuidado que se ha de tener con el material presuntamente infectado de pacientes sospechosos de padecer de ECJ u otra enfermedad por priones, es de suma importancia no solo en la asistencia médica, sino en las investigaciones que se deben realizar al efecto para el esclarecimiento de su fisiopatogenia.
El diagnóstico de las enfermedades por priones siempre entraña dudas en su comienzo, la ECJ tiene una baja incidencia y comparte características comunes con otras causas de demencia, no existen hallazgos significativos en el líquido cefalorraquídeo que puedan orientar al diagnóstico inequívoco y en ocasiones suelen faltar algunos de los elementos que la hacen inconfundible en el orden clínico.9
En el caso presentado, el debut del cuadro con trastornos de conducta, agresividad, alucinaciones visuales y pérdida progresiva de la visión, en un inicio presentó la posibilidad de una encefalitis herpética, y solo luego de aparecer movimientos anormales, con un estado de postración progresiva y cabalgante, con convulsiones tónico-clónicas del hemicuerpo izquierdo, se comenzó a sospechar en la posibilidad de ECJ.
A pesar de lo aparentemente raro del diagnóstico, la ECJ se debe tener en cuenta en la práctica médica diaria y presupone un estado de alerta continuo por la posibilidad de ocurrencia de variantes epidémicas; aun cuando el sistema de vigilancia epidemiológica sea eficaz, estas enfermedades priónicas pueden además afectar tanto a la población en general como incidir, asimismo, en la economía por la aparición de casos en animales en su variante bovina.10
En todo esto puede radicar la importancia principal de este reporte, así como inducir al médico a la investigación no solo de un caso aislado, si no a la investigación en general de las enfermedades por priones que se ciernen como una amenaza para el futuro de la humanidad y pueden representar un peligro potencial a gran escala para la especie humana, pues sus mecanismos de transmisión, su carencia de reconocimiento por el sistema inmune, la aparición de epizootias sumados a la inseguridad de algunos sistemas sanitarios así lo demuestran.11
El diagnóstico patológico se realiza de forma habitual por biopsia cerebral o luego de la necropsia, se incluye en este sentido como recomendaciones en el presente trabajo, el estudio minucioso de otros tejidos extraneurales, como por ejemplo, la biopsia de material extraído de las tonsilas amigdalinas u otros, pues ello aportaría otros medios diagnósticos menos invasivos para el reconocimiento de estas enfermedades priónicas in vivo, en particular la ECJ; además el hallazgo de cambios patológicos extraneurales pudiera arrojar luz sobre el esclarecimiento de la fisiopatogenia y su tratamiento.
En el sentido de su etiología se ha de señalar en este trabajo, la similitud entre algunas enfermedades por priones, especialmente la ECJ con la enfermedad de Alzheimer (EA), y aunque no es objetivo central de la presente publicación, se sugiere en ella la posibilidad de que la EA tenga igualmente un origen priónico o de una partícula infecciosa proteica similar, cuya evolución epidemiológica en cuanto a su comportamiento epidémico y las posibilidades de transmisión mediante inoculación a animales de laboratorio, sus características clínicas en cuanto al desarrollo más o menos lento de una demencia, los conocidos cambios patológicos como la formación de placas amiloides y de ovillos neurofibrilares y los cambios a nivel del núcleo de Meynert12 así como la relación con alteraciones genéticas que ocurren en algunas variantes de ambas entidades pudiera sugerir esta común etiología; investigaciones posteriores demostrarán o no esta hipótesis, de ser así la EA sería la enfermedad priónica más conocida y extendida de todas y el no reconocimiento de esta posible etiología pudiera haber contribuido al desarrollo de la epidemia silenciosa que ha significado desde sus inicios, así como también que sus cambios patológicos se expresarán en la clínica de forma más florida que las demás enfermedades por priones.
Otra posibilidad igualmente hipotética, sería que el concepto de proteína priónica fuera más extenso que lo que se admite en la actualidad y pudiera incluir a otras formas de proteínas anormales como la proteína precursora del amiloide, presenilina 1 y 2 halladas en la EA. El camino queda abierto a las investigaciones en el terreno experimental.
REFERENCIAS BIBLIOGRÁFICAS
1.Villegas VE, Velandia F, Payán C . Enfermedad de Creutzfeldt Jakob esporádica: estudio clínico, patológico y molecular de un caso. Rev Cienc Salud. 2008;6(3):36-46.
2. Jakob, A. Über eine eigenartige Erkrankung des Zentral-Nervensystems mit bemerkenswertem anatomischem Befunde (spastische pseudosklerotische Encephalomyelopathie mit disseminierenden Degenerationsherden). Dtsch Z Nervenheilk. 1921;70:132-46.
3. Creutzfeldt HG, Gesamte Z. Neurol Psychiatr. 1920;57:1-18.
4. Hadlow WJ. Scrapie and kuru. Lancet. 1959; 289-90.
5. Gajdusek DC, Zigas V. Degenerative disease of the central nervous system in New Guinea: epidemic occurrence of «kuru» in the native population. N Engl J Med. 1957;257:974-8.
6. Hill AF, Desbruslais M, Joiner S. The same prion strain causes vCJD and BSE. Nature. 1997;389:448-50.
7. Zivkovic S, Boada M, López O. Revision de la enfermedad del Creutzfeldt-Jakob y otras enfermedades prionicas. Rev Neurol. 2000;31(12):1171-9.
8. Rivero E, Praderes L J, Zamora R. Enfermedad de Creutzfeldt-Jakob en un paciente con infartos cerebrales múltiples. Rev Cubana Med. 1999; 38(4):284-7.
9. Santos S, Pascual-Millán LF, Escalza-Codina I, Navas-Vinagre, López del Val LJ , Mostacero-Miguel E. Ataxia cerebelosa mioclónica progresiva como manifestación de la enfermedad de Creutzfeldt-Jakob. Rev Neurol. 2003;37:535-8.
10. Pacífico C, Galotta JM. Enfermedades por priones. Revista de Ciencias Agrarias y Tecnología de los Alimentos. 2002; Vol. 20.
11.Watts JC , Balachandran A, Westaway D. The Expanding Universe of Prion Diseases. PLoS Pathogens. March 2006;2(3)e26. Disponible en: http://www.plospathogens.org/article/info:doi%2F10.1371%2Fjournal.ppat.0020026
12. Cartier L, Verdugo R, Vergara C, Galvez S. The nucleus basalis of Meynert in 20 definite cases of Creutzfeldt-Jakob disease. Journal of Neurology, Neurosurgery, and Psychiatry. 1989;52:304-9.
Recibido: 24 de febrero de 2009.
Aprobado: 28 de septiembre de 2009.
Dr. Edmundo Rivero Arias. Hospital Clínico Quirúrgico "Hermanos Ameijeiras". San Lázaro # 701 esq. Belascoaín. Centro Habana, Ciudad de La Habana, Cuba. Código postal 10300. Telf. 876 1435. Correo electrónico:erivero@infomed.sld.cu

