










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Medicina
versión impresa ISSN 0034-7523
Rev cubana med vol.50 no.3 Ciudad de la Habana jul.-set. 2011
PRESENTACIÓN DE CASOS
Presentación de un caso clínico de atrofia multisistémica y actualización de criterios diagnósticos
Multisystem atrophy and diagnostic criteria updating: A clinical case presentation
Dr. Alexis Soto Lavastida, Dra. Gloria Lara Fernández, Dr. Enrique Michel Esteban, Dr. Juan Carlos Llibre Guerra
Instituto de Neurología y Neurocirugía "Dr. José Rafael Estrada". La Habana, Cuba.
RESUMEN
La atrofia multisistèmica constituye un trastorno neurodegenerativo esporádico de etiología no precisada que se caracteriza por parkinsonismo, trastornos cerebelosos, disfunción autonómica y piramidalismo; los hallazgos patológicos comprenden pérdida celular y gliosis en las neuronas estriatonígricas, olivopontocerebelosas y autonómicas; y la presencia de inclusiones intracitoplasmáticas oligodendrogliales y neuronales, ubiquitina, tau y alfasinucleína positivas. Afecta tanto a hombres como a mujeres, con inicio en la sexta década de la vida como promedio y una prevalencia de 4/100 000. Se presentaron los últimos criterios diagnósticos de atrofia multisistémica y el caso clínico de un paciente de 65 años con un cuadro progresivo, de 4 años de evolución, ataxia cerebelosa progresiva, síndrome rígido acinético, disfunción autonómica, signos piramidales y mala respuesta a la levodopa, con imágenes de resonancia magnética que muestran atrofia de vermis, hemisferios cerebelosos, tallo cerebral (puente) e hipointensidad de ambas regiones putaminales en t2. Se concluyó el caso con el diagnóstico de atrofia multisistémica tipo C.
Palabras clave: Atrofia multisistémica, degeneración estrionígrica, atrofia olivopontocerebelosa, síndrome de Shy Drager, parkinsonismo, alfa sinucleinopatías.
ABSTRACT
The multisystem atrophy is a sporadic neurodegenerative disorder of unknown origin characterized by parkinsonism, cerebellar disorders, autonomic dysfunction and pyramidal disease, provoked by a cellular loss and gliosis in the nigrostriatal, olivopontocerebellar and autonomic neurons and the presence of oligodendroglia and neuronal intracytoplasmic positive inclusions, ubiquitin, tau and alpha-sinuclein affecting men and women starting as average during the sixth decade of life and a prevalence of 4/100 000. The last diagnostic criteria of multisystem atrophy were showed as well as the clinical case of a patient aged 65 with a progressive picture of 4 years of evolution, progressive cerebellar ataxia, a rigid akinetic syndrome autonomic dysfunction, pyramidal signs and a poor response to levodopa with magnetic resonance images showing vermis atrophy, cerebellar hemispheres, cerebral stem (bridge) and hipointensity of both putamen regions in T2. We conclude that case was diagnosed with type C multisystem atrophy.
Key words: Multisystem atrophy, strionigral degeneration, olivopontocerebellar atrophy, Shy Drager's syndrome, parkinsonism, alpha-sinucleinopathies.
INTRODUCCIÓN
En 1969, Graham y Oppenheimer introdujeron el término de atrofia multisistémica para combinar las entidades siguientes: degeneración estriato nigra, atrofia olivopontocerebelosa y el síndrome de Shy Drager, para describir pacientes con un síndrome neurológico caracterizado por parkinsonismo, ataxia cerebelosa y fallo autonómico.1
La atrofia multisistémica (AMS) es una enfermedad neurodegenerativa que se caracteriza por manifestaciones cardinales de disautonomía, parkinsonismo, ataxia cerebelosa y signos piramidales de causa no precisada aún y comienzo en la adultez.2 Históricamente fueron descritas 3 formas clínicas diferentes, primeramente, Dejerine y Thomas3 introdujeron el término de atrofia olivopontocerebelosa para describir una forma esporádica de comienzo tardío con predominio de un síndrome cerebeloso, acompañado también de parkinsonismo y disautonomía, Shy y Drager4 enfatizaron en una insuficiencia neurogénica central autonómica en pacientes con parkinsonismo y síntomas cerebelosos, finalmente, Adams5 describió las características de un parkinsonismo rápidamente progresivo en pacientes con degeneración estrionígrica.
En 1989 fueron descritas por primera vez inclusiones citoplasmáticas en las células gliales en el cerebro de pacientes con AMS, a pesar de la forma clínica de presentación. En largas series de pacientes con otras enfermedades neurodegenerativas no se hallaron dichas inclusiones. La abundante presencia de inclusiones en todos los subtipos clínicos de AMS contribuyó al reconocimiento de la degeneración estrionígrica, el síndrome de Shy-Drager y la atrofia olivopontocerebelosa esporádica, como una entidad única caracterizada por degeneración neuronal multisistémica, con inclusiones oligodendrogliales.6 A finales de la década de los 90, la AMS fue reconocida como una alfa sinucleinopatía junto con la enfermedad de Parkinson, la demencia por cuerpos de Lewy y el fallo autonómico puro.6,7
Paralelamente, se introdujeron criterios diagnósticos, Quinn fue el primero en proponer criterios diagnósticos,7 otros nuevos fueron desarrollados en 19988 y, por último, en el año 2007.9
Aunque la causa es desconocida, este trastorno parece ser el resultado de una disfunción de la alfa sinucleína.10 La neuropatología de la AMS consiste en degeneración de estructuras estrionígricas y olivopontocerebelosas acompañadas de abundantes y distintivas inclusiones gliales citoplasmáticas.11
La incidencia de la AMS no es bien conocida, se estima en el orden de 3/100 000 por año. Frecuentemente es subestimada y subdiagnosticada. La prevalencia es 4,4/100 000. Sin embargo, el 10 % de los pacientes con parkinsonismo tienen AMS y aportan una prevalencia calculada de 16,4/100 000 hab, mucho mayor que las estimadas en estudios de base poblacional.2,6,12-17
Afecta de forma ligeramente superior a los hombres (1.3:1) y progresa inexorablemente con una supervivencia media de 6 a 9 años.2,12
Teniendo en cuenta las anormalidades motoras, el déficit cerebeloso se desarrolla en aproximadamente 20 % de los pacientes, el 80 % restante se presentan inicialmente con manifestaciones parkinsonianas.10,15
CASO CLÍNICO
Paciente masculino de 65 años, raza blanca, diestro, con antecedentes personales de hipertensión arterial e hiperplasia prostática benigna, no se recogen antecedentes patológicos familiares de enfermedad neurológica alguna. Ingresa por trastornos de la marcha dados por inestabilidad y trastorno del equilibrio, disartria, disfagia, episodios de risa y llanto inmotivados, depresión, caídas frecuentes, disfunción sexual eréctil, incontinencia urinaria, constipación, apnea de sueño, estridor laríngeo y trastornos de la conducta del sueño REM. Curso progresivo de 4 años de evolución. Al ingreso se constata facies hipomímica, disminución de la tasa de parpadeo (10 parpadeos en 1 min), anterocollis, disartria mixta (cerebelosaespástica) hipofonía, marcha a pequeños pasos y atáxica, con aumento de la base de sustentación, imposibilidad de la marcha en tándem, dificultad para los giros, signo de Romberg, dismetría, discronometría, disdiadacocinesia, temblor postural de miembros superiores e intencional de los 4 miembros, además de hipobradicinesia generalizada, pérdida de los reflejos de reequilibración y enderezamiento, rigidez en tubo de plomo, hiperreflexia osteotendinosa generalizada con aumento del área de provocación, reflejo palmomentoniano y policomentoniano, signo de Meyerson y Babinski bilateral, hipotensión ortostática, hipoestesia e hipopalestesia distal de las extremidades. Se constató estridor laríngeo y apnea durante el sueño, no se demostró daño cognitivo.
Exámenes complementarios
- Tomografía computarizada de cráneo simple: Atrofia cortical frontotemporal, cerebelosa.
- Imágenes de RM: Atrofia de vermis, hemisferios cerebelosos y puente (figs. 1 y 2), hipointensidad de ambas regiones putaminales en T2 y presencia de una línea hiperintensa en el borde lateral del putamen (signo de la "hendidura putaminal") (figs. 3 y 4).
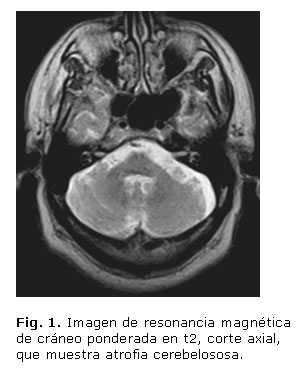
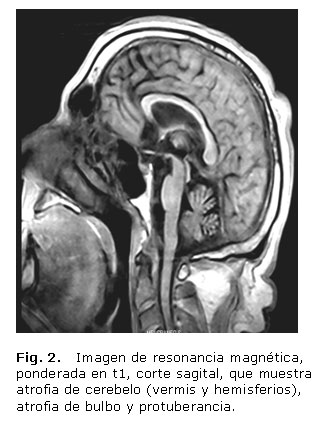
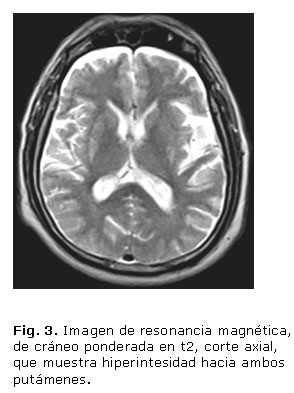
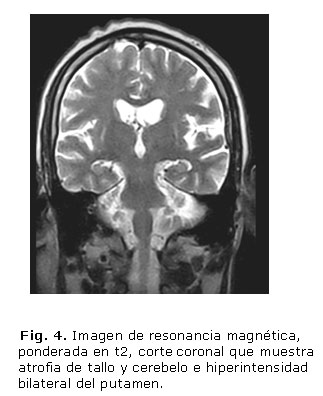
- Prueba urodinámica: Compatible con vejiga hiperrefléctica.
- Electromiograma (EMG) de esfínter anal externo: Signos de desnervación, reinervación crónica.
- Estudios de conducción nerviosa: Neuropatía axonal ligera en miembros inferiores.
Existen síntomas y signos clínicos que pueden incrementar la sospecha de AMS y no de enfermedad de Parkinson (red flags), como se muestra en el cuadro 1.

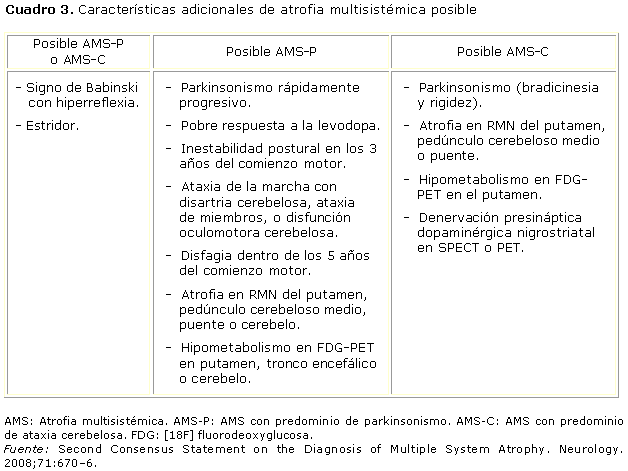
En los cuadros 2 y 3 se muestran los criterios diagnósticos de AMS generados por el Second consensus statement on the diagnosis of multiple system atrophy.

En el caso que aquí presentamos, evolutivamente se observó pobre respuesta a la levodopa. Se concluyó con el diagnóstico de atrofia multisistémica tipo C.
DISCUSIÓN
Existen síntomas y signos clínicos que pueden incrementar la sospecha de AMS y no de enfermedad de Parkinson (red flags). El paciente presentado tiene varios elementos que llaman poderosamente la atención: curso de la enfermedad rápidamente progresivo, sin antecedentes familiares de interés patológico, un síndrome cerebeloso tanto vermiano como hemisférico, síndrome parkinsoniano bilateral simétrico con distonía del cuello, pobre respuesta a la l-dopa, ausencia de temblor de reposo, trastorno de la mecánica ventilatoria (estridor laríngeo), disautonomía prominente (incontinencia urinaria, fallo autonómico), disartria precoz y signos piramidales.
Lo anteriormente observado nos obliga a descartar no solo la forma idiopática del síndrome parkinsoniano (enfermedad de Parkinson) sino también la heredodegenerativa y, al no tener antecedentes por los cuales podamos pensar en una causa secundaria, nos queda solamente la categoría etiológica de las formas atípicas del síndrome.
Sin demencia, trastorno de la conducta, parálisis vertical de la mirada, apraxia, fenómeno de la mano ajena, trastorno sensitivo cortical, mioclonía y asimetría destacada del síndrome parkinsoniano, se sugiere el diagnóstico de atrofia multisistémica.
Como lo refiere la literatura revisada, el caso clínico que se reporta tiene parkinsonismo rígido acinético bilateral asimétrico, este elemento es predictor de pobre supervivencia, el temblor postural es frecuente, pero es rara la presencia del clásico temblor de reposo en cuentamonedas.1,2,5,10,15 El predominio del síndrome cerebeloso, ataxia de miembros y de la marcha, y la disartria cerebelosa hace que diagnostiquemos la AMS tipo cerebelosa .2,12 Los trastornos cerebelosos y parkinsonianos están frecuentemente combinados. La mitad de los pacientes tienen signos piramidales (hiperreflexia osteotendinosa y signo de Babinski) como se pudo constatar en este paciente.7,10,15
La parálisis bilateral de las cuerdas vocales en el paciente es la consecuencia del estridor inspiratorio aunque junto con la insuficiencia respiratoria pueden ser los síntomas primarios de presentación de la AMS.2,7.10,14
Según reportan los artículos revisados, la mayoría de los pacientes con AMS puede presentar pobre respuesta al tratamiento con levodopa, como ocurrió con el paciente que se presenta, se ha reportado que 30 % de los pacientes tienen buena respuesta a la levodopa aunque nunca mantenida en el tiempo como en la enfermedad de Parkinson.2,10
En la revisión del tema, hallamos que la disautonomía se manifiestò en 41 % de los pacientes inicialmente y evolutivamente, en el 97 %. La hipotensión postural sintomática se presentó en 68 % de los pacientes.12 La impotencia es casi universal en los hombres y en las mujeres predomina la incontinencia. Esa afirmación, en cuanto a los síntomas disatonómicos, fue corroborada en el paciente aquí reportado.2,7,10.18
Como se constata en otros casos clínicos reportados, la enfermedad se inicia por síntomas de un solo sistema lo que hace difícil el diagnóstico correcto y precoz, pero la naturaleza progresiva y la participación gradual de otros sistemas inicialmente no afectados facilitan el diagnóstico. Los pacientes en los que inicialmente se hallan manifestaciones extrapiramidales, comúnmente progresan para desarrollar disfunción autonómica, trastornos cerebelosos o ambos, de forma similar los síntomas y signos en otros tipos de AMS muchas veces evolucionan a otros sistemas fuera de los afectados en la evaluación inicial, como sucedió con el paciente reportado.2,7,10,14-15,19.
Aunque en la RM cerebral, con frecuencia, aparecen alteraciones que incluyen atrofia putaminal, hipointensidad del putamen (relativa al globo pálido) en imágenes en t2, así como cambio de señal en forma de hendidura (slit-like signal) en el margen posterolateral del putamen, esta intensidad anormal puede ser asimétrica, las anormalidades infratentoriales incluyen atrofia cerebelosa y del tronco encefálico, así como cambio de señal en la protuberancia (signo de la cruz caliente) y el pedúnculo cerebeloso medio. La base pontina y el pedúnculo cerebeloso medio pueden mostrar hiperintensidad en t2, que indica degeneración y desmielinización.2,6,12 El caso clínico que reportamos presenta atrofia de cerebelo (vermiana como hemisférica), de tallo cerebral (bulbar y pontina) y señal hiperintensa bilateral del putamen, elementos que apoyan el diagnóstico, la atrofia cerebelosa muy prominente es la que apoya el diagnóstico de AMS tipo C.
La manifiestación de la pérdida neuronal en el núcleo de Onuf, mediante los estudios neurofisiológicos (electromiograma de esfínter anal), apoya el pensamiento clínico. Según se afirma en la literatura revisada acerca de este tema, en el electromiograma se destacan elementos como desnervación y reinervación crónica, los cuales fueron encontrados en el estudio neurofisiológico de nuestro paciente. No obstante, para apoyar el diagnóstico se puede utilizar además el electromiograma del esfínter uretral.2,6,12
Para tener una aproximación clínica al diagnóstico del paciente reportado seguimos los criterios clínicos reportados por la literatura, del Second consensus statement on the diagnosis of multiple system atrophy.9
De forma similar al primer consenso, el segundo determinó que la AMS debería ser clasificada en 3 grupos:
· AMS definida.
· AMS probable.
· AMS posible.
Cuando predominan las manifestaciones parkinsonianas se continúa diagnosticando como atrofia multisistémica con predominio de parkinsonismo (AMS-P) y cuando predomina la ataxia cerebelosa, como atrofia multisistémica con predominio de ataxia cerebelosa (AMS-C).
El caso clínico que se reporta, según el segundo consenso, entra en la categoría de AMS tipo C probable por aparecer en un paciente con más de 30 años, ser esporádica y no hereditaria, tener fallo autonómico, incontinencia urinaria, parkinsonismo con pobre respuesta a la l-dopa, síndrome cerebeloso y piramidalismo.
REFERENCIAS BIBLIOGRÁFICAS
1. Graham JG, Oppenheimer DR. Orthostatic hypotension and nicotine sensitivity in a case of multiple system atrophy. J Neurol Neurosurg Psychiatry. 1969;32: 28-34.
2. Bhidayasiri R, Ling H. Multiple System Atrophy. T Neurologist. 2008;14(4):224-37.
3. Dejerine J, Thomas A. Látrophie olivo-pontocerebelleuse. Nouv Iconogr Salpetriere. 1900;13:300-70.
4. Shy GM, Drager GA. A neurological syndrome associated with orthostatic hypotension. Arch Neurol. 1960;2:511-27.
5. Adams RD, Van Bogaert P, Eecken VD. Degeneration nigro-striees et cerebello-nigro-striees. Phychiatr Neurol. 1961;142:219-59.
6. Jankovic J, Tolosa E. Multiple System Atrophy. En: Parkinson's Disease & Movement Disorders. 5th ed. Philadelphia: Lippincott Williams & Wilkins; 2007.176-85.
7. Quinn NP. How to diagnose multiple system atrophy. Mov Disord. 2005;20(suppl 12):S5-S10.
8. Gilman S, Low PA, Quinn N, Albanese A, Ben-Shlomo Y, Fowler CJ, et al.Consensus statement on the diagnosis of multiple system atrophy. J Auton Nerv Syst. 1998 Dec 11;74(2-3):189-92.
9. Gilman S, Wenning GK, Low PA, Brooks DJ, Mathias CJ, Trojanowski JQ, et al. Second consensus statement on the diagnosis of multiple system atrophy. Neurology. 2008;71:670-6.
10. Kaufmann H, Goldstein D. Autonomic failure in neurodegenerative disorders. Continuum Lifelong Learning Neurol. 2007;13(6):111-42.
11. Ozawa T, Paviour D, Quinn NP, Josephs KA, Sangha H, Kilford L, et al. The spectrum of pathological involvement of the striatonigral and olivopontocerebellar systems in multiple system atrophy: clinicopathological correlations. Brain. 2004;127:2657-71.
12. Kaufmann H. Disautonomías más comunes. Rev Neurol. 2003;36(1):93-6.
13. Wenning G, Geser F, Poewe W. Therapeutic Strategies in Multiple System Atrophy. Mov Disord. 2005;20(12):S67-S76.
14. Köllensperger M, Geser F, Seppi K, Stampfer-Kountchev M, Sawires M, Scherfler C, et al, on behalf of the European MSA Study Group (EMSA-SG). Red Flags for Multiple System Atrophy. Mov Disord. 2008;23(8):1093-9.
15. Wenning GK, Ben Shlomo Y, Magalha~es M, Danie S, Quinn NP. Clinical features and natural history of multiple system atrophy. An analysis of 100 cases. Brain. 1994;117(pt 4):835-45.
16. Geser F, Wenning GK, Seppi K, Stampfer-Kountvhev M, Scherfler C, Sawires M, et al. Progression of multiple system atrophy (MSA): a prospective natural history study by the European MSA Study Group (EMSA SG). Mov Disord. 2006;21:179-86.
17. Geser F, Seppi K, Stampfer-Kountchev M, Köllensperger M, Diem A, Ndavisaba JP, et al. The European Multiple System Atrophy-Study Group (EMSA-SG). J Neural Transm. 2005;112:1677-86.
18. Kirchhof K, Apostolidis AN, Mathias CJ, Fowler CJ. Erectile and urinary dysfunction may be the presenting features in patients with multiple system atrophy: a retrospective study. Int J Impot Res. 2003;15:293-8.
19. Watanabe H, Saito Y, Terao S, Ando T, Kachi T, Mukai E, et al. Progression and prognosis in multiple system atrophy: an analysis of 230 Japanese patients. Brain. 2002;125:1070-83.
Recibido: 24 de julio de 2010.
Aprobado: 17 de marzo de 2011.
Dr. Alexis Soto Lavastida. Instituto de Neurología y Neurocirugía "Dr. José Rafael Estrada", calle 29 y D, El Vedado. La Habana, Cuba.


{kind=link}