










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Medicina
versión impresa ISSN 0034-7523
Rev cubana med vol.52 no.1 Ciudad de la Habana ene.-mar. 2013
PRESENTACIÓN DE CASO
Síndrome de POEMS
Poems syndrome
Dra. Karen Valdés Álvarez, Dr. Luis Senra Armas, Dra. Gengly Aguilar Linares
Hospital Clinicoquirúrgico "Hermanos Ameijeiras". La Habana, Cuba.
RESUMEN
El síndrome de POEMS es un trastorno paraneoplásico asociado a discrasia de células plasmáticas que reúne, entre sus criterios diagnósticos mayores, la presencia de polineuropatía y de gammapatía monoclonal, entre otras como esclerosis ósea y/o cutánea. Se reportó un caso que reúne las características dominantes del síndrome.
Palabras clave: síndrome POEMS, polineuropatía, esclerosis cutánea, esclerosis ósea.
ABSTRACT
POEMS syndrome is a paraneoplastic disorder associated with plasma cell dyscrasia which joins among itsmajor diagnostic criteria, the presence of polyneuropathy and monoclonal gammopathy, such as bone and / or skin sclerosis. a case that meets the key features of the syndrome is reported.
Key words: POEMS syndrome, polyneuropathy, cutaneous sclerosis, bone sclerosis.
INTRODUCCIÓN
El síndrome de POEMS es un trastorno paraneoplásico asociado a discrasia subyacente de células plasmáticas.1 Este acrónimo (POEMS), sugerido por Bardwik en 1980,2 engloba las características dominantes del síndrome, a saber: polineuropatía, organomegalia, endocrinopatía, proteína M y alteraciones cutáneas. Otros trastornos relacionados con este síndrome, incluidos entre los criterios diagnósticos, son la enfermedad de Castleman, la elevación del factor de crecimiento del endotelio vascular, el papiledema y la trombocitosis/policitemia.
La polineuropatía asociada al POEMS es de tipo desmielinizante, la cual se considera un criterio diagnóstico mayor y constituye el trastorno inicial en aproximadamente 50 % de los casos.3 Las manifestaciones cutáneas incluyen hiperpigmentación, engrosamiento dérmico, hirsutismo e hipertricosis, por lo que se plantea un diagnóstico diferencial con enfermedades del tejido conectivo.1,4
En esta oportunidad se presenta una paciente con polineuropatía severa y extensas lesiones escleróticas en piel, a la cual se le realizó el diagnóstico de POEMS tardíamente.
PRESENTACIÓN DEL CASO
Mujer de 52 años de edad, raza blanca, quien hace 3 años comenzó a padecer dolor punzante en ambos miembros inferiores, con predominio distal, que fue incrementándose de forma progresiva asociado a calambres y adormecimiento, y que luego se extendió a los miembros superiores. Evolutivamente, presentó disminución de la fuerza muscular y compromiso de la marcha. Dicho cuadro se interpretó como una polineuropatía desmielinizante inflamatoria crónica (CIDP) tratada con corticoesteroides e inmunoglobulina intravenosa (IVIG), pero no se logró una respuesta terapéutica satisfactoria.
Dos años después del inicio de los síntomas aparecieron manifestaciones cutáneas dadas por: coloración rojoviolácea, induración y engrosamiento de la piel de la cara, cuello, antebrazos, manos y pies, dificultad en la apertura de la boca y afinamiento de los dedos de las manos. Tres meses antes del ingreso se sumó a este cuadro, disnea a los grandes y medianos esfuerzos.
En la exploración física, al momento del ingreso, se encontró: induración y engrosamiento cutáneo en cara, cuello, antebrazos, manos y pies, facies de ratón, ausencia de pliegues cutáneos, limitación de la apertura de la boca, enrojecimiento cutáneo en cuello, dorso y antebrazos, dedos afinados y flexión permanente del 4to. y 5to. dedos de ambas manos (Figs. 1, 2 y 3). Se auscultaron crepitantes finos en la base pulmonar derecha, reforzamiento del 2do. ruido en el foco pulmonar y hepatosplenomegalia.
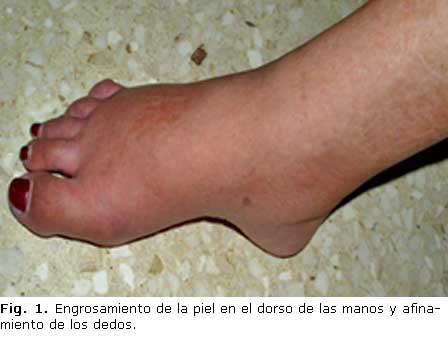
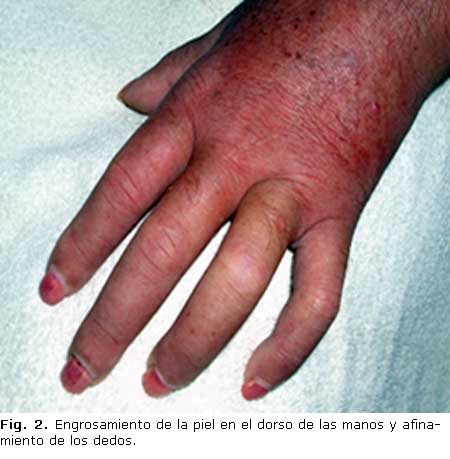

.
A la exploración neurológica se constató marcha en estepaje, hiperpatía al nivel del pulpejo de todos los dedos, hipopalestesia, hipobatiestesia, hipotrofia muscular distal y crural; disminución de la fuerza muscular con predominio distal y crural, y abolición de todos los reflejos osteotendinosos.
En los exámenes complementarios realizados se encontró hemoglobina, química sanguínea y eritrosedimentación, normales y anticuerpos antinucleares, negativos. Un pequeño componente M fue hallado en la electroforesis de proteínas (banda pequeña en gamma, 18,8 g/L) y en la inmunofijación se demostró un pico monoclonal de IgG lambda.
Los exámenes endocrinológicos mostraron disminución en los niveles de testosterona, con un valor de 0,25 nmol/L (VR 0,22 2,9 nmol/L) y la prolactina elevada con valor de 602,5 µUI/mL (VR 71,1 511,99 µUI/mL).
La neuroconducción y la electromiografía mostraron hallazgos compatibles con polineuropatía desmielinizante y el líquido cefalorraquídeo resultó con proteínas elevadas, en el rango de 1,260 g/L, sin la presencia de células.
En los estudios imaginológicos, el ultrasonido y la tomografía computarizada mostraron hepatosplenomegalia sin mostrar adenopatías. Además, se precisaron lesiones osteoescleróticas en columna dorsal, esternón y costillas (Fig. 4).

Se encontró un patrón restrictivo en las pruebas de función respiratoria y una hipertensión pulmonar ligera en el ecocardiograma transtorácico.
En el medulograma se encontró una infiltración por células plasmáticas menor de 10 %.
DISCUSIÓN
El síndrome de POEMS también conocido como síndrome de Crow Fukase o síndrome de Takatsuki es un trastorno asociado a una discrasia de células plasmáticas subyacente. El pico de incidencia está entre la 5ta. y la 6ta. décadas de la vida y afecta con mayor frecuencia a hombres (2,5:1).5
La mayoría de los pacientes tienen un mieloma osteoesclerótico. Su causa es compleja y aún no bien establecida. La elevación del factor de crecimiento endotelial vascular y la presencia de citoquinas proinflamatorias son el sello distintivo de este trastorno, en estrecha relación con su patogenia.1,5
En diferentes series publicadas pocos casos reunían las 5 características originales del síndrome, por lo que se definieron nuevos criterios por la Clínica Mayo, basados en 5 criterios mayores (2 de ellos obligatorios) y 6 criterios menores. Para realizar el diagnóstico se requiere al menos 3 criterios mayores y un criterio menor.1,5,6
La principal característica clínica de este síndrome es una polineuropatía progresiva crónica con predominio motor prácticamente presente en todos los pacientes. Los síntomas comienzan en los pies de forma simétrica y consisten en parestesias, adormecimiento y enfriamiento. Las manifestaciones motoras siguen a los síntomas sensitivos y predominarán a posteriori. De las regiones distales continúan hacia la parte proximal de los miembros inferiores y después afectan a los miembros superiores. La debilidad severa ocurre en más de la mitad de los pacientes y ocasiona imposibilidad de subir escaleras o incorporarse de la posición sentada. Hay disminución o ausencia de los reflejos osteotendinosos. Los estudios de conducción nerviosa y electromiografía demuestran una polineuropatía con prominente desmielinización tanto como degeneración axonal, lo cual puede ser similar a los hallazgos de pacientes con CIDP. Sin embargo, la CIDP se caracteriza por involucrar tanto las regiones proximales como las distales y tiene mejor respuesta al tratamiento con inmunoglobulinas y esteroides.3,4 La polineuropatía encontrada en la paciente cumplía con todas estas características y precedieron en 3 años a la aparición del resto de los síntomas.
La organomegalia generalmente involucra al hígado y ocurre sin características patológicas. La esplenomegalia y los linfonodos representan las características de la enfermedad de Castleman, asociada de 11 % a 33 % de los casos. En el presente caso predominaba la hepatomegalia sobre la esplenomegalia y no se encontraron adenomegalias para apoyar el diagnóstico de Castleman.7
Otro criterio mayor es la presencia de proteína M cuyo tamaño electroforético es pequeño (media 1,1 g/dL), raramente superior a 3,0 g/dL. La proteína M es, por lo general, IgG o IgA, casi siempre de tipo lambda y frecuentemente tiene solo 5 % o menos de células plasmáticas en medula ósea.8 En el caso presentado fue la curva electroforética la que arrojó el elemento de monoclonalidad gamma dado que la cuantificación de proteínas se encontraba dentro de límites normales.
Las lesiones osteoescleróticas ocurren en, aproximadamente, 95 % de los pacientes y pueden crear confusión con otras lesiones benignas del hueso como fibromas no osificados y displasias fibrosas.8 Pueden encontrarse también lesiones osteolíticas y combinadas, osteolíticas con reborde osteoblástico.
La endocrinopatía es una característica central pero pobremente comprendida. En una serie reciente de la Clínica Mayo, aproximadamente, al 84 % de los pacientes se les reconoció una endocrinopatía con hipogonadismo como la anormalidad endocrina más común, seguida de anormalidades tiroideas, anormalidades en el metabolismo de la glucosa y por último, insuficiencia adrenal.9
Múltiples cambios dermatológicos se han asociado con el síndrome de POEMS. Los más comunes incluyen hiperpigmentación, engrosamiento de la piel, cambios esclerodérmicos e hipertricosis. En la paciente estudiada, las lesiones cutáneas eran extensas y predominó la esclerosis.1,4,10
Las manifestaciones pulmonares son variadas, incluyen hipertensión pulmonar y enfermedad pulmonar restrictiva, como en este caso. En una serie de 20 pacientes con POEMS y seguimiento por 10 años, 25 % manifestó hipertensión pulmonar. Otras anormalidades incluyen, alteración de la función respiratoria neuromuscular y la capacidad de difusión del monóxido de carbono comprometida.11
El pronóstico de los pacientes con este síndrome es pobre, se considera que la supervivencia es de 12 a 33 meses.8 La terapia médica combina altas dosis de corticosteroides y quimioterapia (melfalán)1,4 con el trasplante de progenitores hematopoyéticos obtenidos de sangre periférica.12 La lesión osteosclerótica solitaria requiere radioterapia local.
REFERENCIAS BIBLIOGRÁFICAS
1. Dispenzieri A. POEMS Syndrome. Blood Reviews. 2007;21:285-99.
2. Bardwick PA, Zvaifler NJ, Gill GN, Newman D, Greenway DL, Resnick DL. Plasma cell dyscrasia with polyneuropathy, organomegaly, endocrinopathy, M protein, and skin changes: the POEMS syndrome. Report on two cases and a review of the literature. Medicine. 1980;59:311-22.
3. Nasu S, Misawa S, Sekiguchi Y. Different neurological and physiological profiles in POEMS syndrome and chronic inflammatory demyelinating polyneuropathy. J Neurol Neurosurg Psychiatry. May. 2012;83(5):476-9.
4. Valenzuela F, Gajardo MF, Gutiérrez DM, Herrera PA. Síndrome de POEMS. Piel. Formación continuada en dermatología. 2012;27:453-5.
5. Dispenzieri A, Kyle R, Lacy M, Rajkumar S, Therneau T. POEMS syndrome definitions and long term outcome. Blood. 2003;101:2496-506.
6. Kuwabara S, Dispenzieri A, Arimura K, Misawa S. Treatment for POEMS (polyneuropathy, organomegaly, endocrinopathy, M-protein, and skin changes) syndrome. Cochrane Database Syst Rev 2008:CD006828. [PubMed: 18843731].
7. Cheng E, Chee A, Dispenzieri A. Amyloidosis and POEMS symdrome. Expert Opin Pharmacother. Jun. 2010;11(9):1501-14.
8. Soubrier MJ, Dubost JJ, Sauvezie BJ, French Study Group on POEMS Syndrome. POEMS syndrome: a study of 25 cases and a review of the literature. Am J Med. 1994;97:543-53. [PubMed: 7985714.
9. Gandhi GY, Basu R, Dispenzieri A, Basu A, Montori VM, Brennan MD. Endocrinopathy in POEMS syndrome: the Mayo Clinic experience. Mayo Clin Proc. 2007;82:836-42. [PubMed: 17605964].
10. Misri R, Kharkar V, Dandale A, Patel V, Mahajan S, Khopkar U. Multiple capillary hemangiomas: A distinctive lesion of multicentric Castleman's disease and POEMS syndrome. Indian J Dermatol Venereol Leprol. 2008;74:36-46.
11. Allam JS, Kennedy C, Aksamit T, Dispenzieri A. Pulmonary Manifestation in Patients with POEMS syndrome: a Retrospective Review of 137 patients. Chest. 2008;133;969-74.
12. Dispenzieri A, Lacy MQ, Hayman SR. Peripheral blood stem cell transplant for POEMS syndrome is associated with high rates of engraftment syndrome. Eur J Haematol. 2008;80:397-406. [PubMed: 18221391].
Recibido: 20 de diciembre de 2012.
Aprobado: 7 de enero de 2013.
Dra. Karen Valdés Álvarez. Hospital Clinicoquirúrgico "Hermanos Ameijeiras", San Lázaro No. 701 entre Belascoaín y Marqués González, Centro Habana, La Habana, Cuba. CP 10 300.
