










 Serviços customizados
Serviços customizados Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
PermalinkIntroducción
El Síndrome de Marfán (SM) es un trastorno sistémico del tejido conectivo, causado por mutaciones en las proteínas de la matriz extracelular fibrilina 1 (FBN1), caracterizada por una combinación variable de manifestaciones cardiovasculares, musculo-esqueléticas, oftalmológicas y pulmonares. Este afecta por igual a ambos sexos y presenta una prevalencia que se estima que sea de 2 a 3 casos por cada 10 000 individuos.1
En el año 1896 el pediatra francés Bernard Antoine Marfán, profesor de la Universidad de París, presentó el caso de una niña de cinco años de edad, Gabrielle P, cuyos dedos y brazos eran, desproporcionalmente, largos unidos a delgadez ósea. Marfán utilizó el término de “dedos de araña” (aracnodactilia) y dolicoestenomelia (miembros largos). Seis décadas después en el año 1955 el Dr. Víctor Kusick, médico y genetista del hospital de Johns Hopkins describió, nuevamente, el Síndrome de Marfán.1,2
Este síndrome se caracteriza por alargamiento de las extremidades, hiperlaxitud articular, dolicocefalia, alteraciones odontológicas y cardiovasculares con lesiones valvulares, fundamentalmente, en las válvulas mitrales. Aunque el diagnóstico es clínico basado en una serie de criterios médicos y genéticos denominados nosología de Ghent, se pueden realizar estudios moleculares para identificar la mutación del gen implicado en esta entidad.
Se hace necesario realizar un correcto diagnóstico diferencial con otras entidades que presentan algunas manifestaciones clínicas semejantes y precisar el diagnóstico definitivo debido a la presencia de complicaciones a mediano o largo plazo que pueden deteriorar la calidad de vida de los enfermos.
Se realiza la siguiente presentación teniendo en cuenta la baja morbilidad de esta enfermedad y la escasez de reportes sobre ella. Se presentan dos casos con el objetivo de describir el diagnóstico de los pacientes con síndrome de Marfán.
Caso clínico
Caso clínico 1
El motivo de la consulta fue el decaimiento. Historia de la enfermedad actual: Paciente ALA masculino de 17 años de edad, color de la piel blanca, con antecedentes referidos de salud, hasta hace un año que comienza con aumento considerable de su talla, alargamiento de las extremidades y de las falanges, con afinamiento de estas últimas y aumento de su flexibilidad. Seis meses después presentó palpitaciones, falta de aire exacerbada a los grandes o continuos esfuerzos, vértigos, tos y decaimiento. Acude a consulta médica y luego de realizar un minucioso examen físico se decide indicar exámenes complementarios para precisar el diagnóstico.
Antecedentes patológicos personales: No refiere.
Antecedentes patológicos familiares: Madre viva, sana. Padre vivo con HTA.
Operaciones, traumatismos, transfusiones y hábitos tóxicos: No refiere.
Datos positivos al examen físico: Hiperemia gingival con apelotonamiento dental. SOMA: Dolicocefalia, disminución del panículo adiposo, alargamiento de las extremidades y las falanges, escoliosis y pie plano (Fig. 1 y 2).
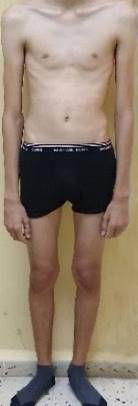
Sistema respiratorio: Tórax enfisematoso, expansibilidad torácica, vibraciones vocales y murmullo vesicular disminuido, hipersonoridad pulmonar.
Sistema cardiovascular: Latido de la punta desplazado hacia la izquierda de la línea media clavicular, presencia de latido epigástrico, ruidos cardíacos taquicárdicos. Se ausculta soplo holosistólico, III-IV/VI, suave en foco mitral con irradiación a la axila.
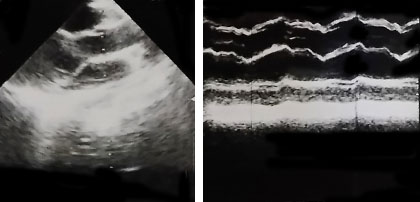
Exámenes complementarios: Hb 11,2 g/L, LCN 6,0 x 109/L, creatinina 70 mmol/L, ácido úrico 175 mmol/L, glicemia 3,8 mmol/L, colesterol 2,0 mmol/L, EKG: Taquicardia sinusal, Rx de tórax PA: Signos de enfisema pulmonar y aumento del índice cardiotorácico a expensas del ventrículo izquierdo, Ecocardiograma (11-02-2018): Valva anterior de la mitral elongada, con prolapso ligero hacia atrio izquierdo e insuficiencia mitral. (Fig. 3).
Caso clínico 2
El motivo de la consulta fue la fatiga. Historia de enfermedad actual: Paciente LEA, masculino de 20 años de edad, color de la piel blanca, con antecedentes referidos de salud. Hace cuatro años comienza con fatiga a los esfuerzos, debilidad muscular, decaimiento, déficit visual y alargamiento de las falanges y de la longitud de sus extremidades. Es remitido a consulta Medicina Interna para precisar su diagnóstico.
Antecedentes patológicos personales: No refiere.
Antecedentes patológicos familiares: Madre viva, sana. Padre sano.
Operaciones, traumatismos y transfusiones: No refiere.
Hábitos tóxicos: No refiere.
Datos positivos al examen físico:
Diastema en la arcada dentaria superior.
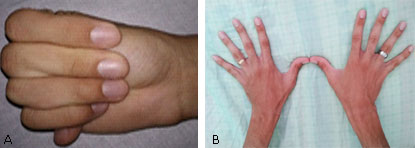
SOMA: Escoliosis, pie plano. Disminución del panículo adiposo, dolicocefalia. Paladar ojival y diastema (Fig. 4). Alargamiento de las extremidades y las falanges, con aproximación del 1er y 5to dedo al agarrar la muñeca (Fig. 5).
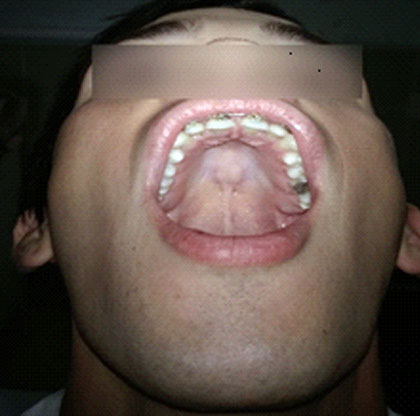
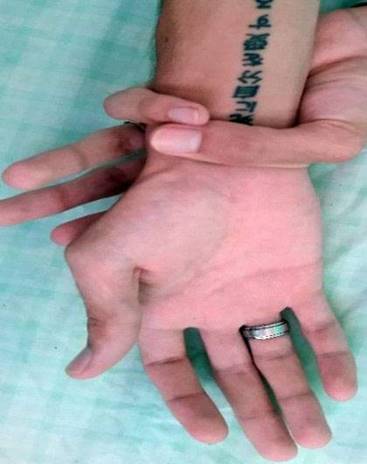
Sistema cardiovascular: Latido de la punta desplazado hacia la izquierda de la línea media clavicular, ruidos cardíacos taquicárdicos. Se ausculta soplo holosistólico, III/VI, suave en foco mitral con irradiación hacia el ápex.
Exámenes complementarios: Hb 12,1 g/L, LCN 5,0 x 109/L, creatinina 62 mmol mmol/L, ácido úrico 210 mmol/L, glicemia 3.6 mmol/L, colesterol 2.8 mmol/L.
EKG: Bradicardia sinusal.
Rx de tórax PA: Aumento de la transparencia pulmonar.
Ecocardiograma (12-11-15): Regurgitación pulmonar ligera, prolapso de valva anterior mitral con regurgitación mitral. Insuficiencia pulmonar, tricuspídea y mitral ligeras.
Discusión
El Síndrome de Marfán es un trastorno sistémico del tejido conectivo, de tipo autosómico dominante, causado en la mayoría de los casos por mutaciones en la proteína de la matriz extracelular fibrilina1 (FBN1). La fibrilina 1 es una glicoproteína predominante en la matriz extracelular, codificada solo por un gen FBN1 constituido por 65 exones, localizado en el cromosoma 15q21.2,3 Esta es un componente importante de los tejidos conectivos elásticos y no elásticos, y es la principal proteína de un grupo de microfibrillas del tejido conectivo que son esenciales para una normal fibrilogénesis elástica.
En cultivos de fibroblastos se observa que los afectados tienen, por lo menos, un 50 % de ausencia de microfibrillas en relación con los no afectados.2,3,4 Se describen más de 200 mutaciones para este gen causantes del SM. Se han identificado, además, nuevas formas relacionadas de la enfermedad, sobre todo secundarias a mutaciones en el gen TGFBR2 localizado en el cromosoma 3, el cual codifica para el receptor del TGF-beta.4 Sin embargo, a pesar de estos avances en la patogénesis del SM, la notable variabilidad fenotípica intrafamiliar e interfamiliar continúa siendo uno de los retos principales de esta entidad.4,5,6
Los síntomas y signos pueden aparecer a cualquier edad y son muy variables de una persona a otra, incluso dentro de una misma familia. Nuestros enfermos se diagnosticaron a los 17 y a los 20 años de edad, los estudios sobre estos casos reportan que no se diagnostican durante la infancia temprana, sino más tardíamente, pues es a partir de la adultez que llaman la atención las características de su biotipo y las manifestaciones clínicas patognomónicas de la afección.1,2,5
En la literatura consultada se definen criterios mayores y menores para el diagnóstico de esta entidad, entre ellos: esqueléticos, cardiovasculares, oculares, pulmonares, neurológicos, genéticos y dermatológicos. Por lo general, la afección esquelética es el primer signo de la enfermedad y puede incluir una dolicoestenomelia, talla grande, aracnodactilia, hipermovilidad articular, escoliosis, protrusión del acetábulo, deformidad torácica con pectus carinatum o excavatum, dolicocefalia e hiperlaxitud articular.5,7,8
Los autores describen que es de vital importancia para el diagnóstico la búsqueda de los signos de Steinberg y Walker-Murdoch, los cuales son avalados como criterios imprescindibles. El primero consiste en la protrusión del pulgar en oposición forzada más allá del borde cubital de la mano y el segundo es evidente al superponerse los dedos pulgar y meñique en más de 1-2 cm al hacer prensión de la muñeca, proximalmente a la apófisis estiloides del radio con la otra mano, ambos signos presentes en nuestros pacientes.7,8
En los casos que presentamos se encontraron estos síntomas y signos excepto el pectus carinatum o excavatum; sin embargo, se detectaron signos clínicos de enfisema pulmonar. Por otra parte, se encontraron alteraciones odontológicas, en el primer caso apelotonamiento dental y paladar ojival; en el segundo caso separación de los incisivos centrales (diastema) y similar lesión del paladar. Numerosos autores hacen referencia a la existencia de alteraciones de la voz y el lenguaje que requieren manejo logopédico;1,8 sin embargo, en nuestros pacientes no se comprobaron estos síntomas.
La afección cardiovascular se caracteriza por dilatación progresiva de la aorta, acompañada de un riesgo elevado de disección aórtica, la dilatación aórtica puede conducir a una insuficiencia de la válvula aórtica e insuficiencia de la mitral, según la literatura consultada se evidenció que el daño valvular más frecuente en estos pacientes es el prolapso de la válvula mitral.9,10 A ambos pacientes se les detectaron lesiones valvulares mitrales y al segundo caso, además, lesiones tricuspídeas y pulmonares.
La afectación oftalmológica lleva a una ectopia o luxación del cristalino. Así mismo, pueden presentarse signos cutáneos (estrías atróficas), riesgo de neumotórax y ectasia dural. No se encontraron estas alteraciones en los enfermos estudiados.1,5
Aunque el diagnóstico de esta entidad es, eminentemente, clínico, se describe la realización de estudios moleculares para la identificación de mutaciones en el gen FBN1, imposibles de realizar en nuestro medio.
Nuestros pacientes presentan vínculos familiares y fueron remitidos del área de salud a consulta, interconsultándose por especialidades de Medicina Interna, Cardiología y Genética. En ambos casos se constataron rasgos clínicos característicos del SM.
Aplicando un correcto método clínico se descartaron otras entidades que son necesarias diferenciar en nuestros casos, entre ellas, la aranodactilia contractual congénita, síndrome de Stickler, síndrome de X frágil,1,5,9 el síndrome de Ehlers-Danlos y el pseudoxantoma elástico.9
Actualmente, el manejo de estos pacientes se realiza teniendo en cuenta medidas de prevención, de rehabilitación, vigilancia y terapéutica oportuna de las complicaciones. El tratamiento con β bloqueadores se recomienda como profiláctico en cualquier paciente con SM y dilatación de la raíz aórtica, ya que puede disminuir su progresión, especialmente en aquellos con un diámetro aórtico menos de 40 mm. Cuando la dilatación aórtica alcanza los 55 mm en adultos o 50 mm en niños se deber realizar cirugía profiláctica con reemplazo de la aorta ascendente y la válvula aortica.1,9,11
Se sugiere no realizar deportes competitivos, de contacto, ejercicios isométricos ni actividades que causen lesión o dolor de las articulaciones. Se debe evitar el consumo de fármacos que estimulen el sistema cardiovascular como los descongestivos. La cafeína puede agravar la tendencia a las arritmias, para pacientes con tendencia a presentar neumotórax se prohíbe el uso de instrumentos de viento o ventilación con presión positiva.12,13
El manejo de estos pacientes incluye medidas de rehabilitación músculo-esqueléticas y psicomotoras. Las deformaciones de la columna vertebral (escoliosis) y del tórax pueden experimentar mejoría mediante sesiones de kinesioterapia y, en ocasiones, mediante el uso de un corsé.8,12,13,14 La kinesioterapia permite además aliviar los dolores articulares, sobre todo de la columna vertebral.
El tratamiento de las complicaciones oculares, cardiovasculares, esqueléticas y dentales, fundamentalmente, debe realizarse oportunamente.14 El personal médico recomienda el empleo de lentes, cirugías del cristalino y de la aorta cuando sean necesarias, así como estabilización de la columna a causa de la escoliosis, corrección con plantillas del pie plano y uso de expansores de paladar.
Los dos enfermos estudiados actualmente tienen estabilidad clínica, reciben tratamiento sintomático y fisioterapia, seguimiento por consulta externa y se realiza vigilancia de futuras complicaciones.
Es importante la divulgación y estudio de estos casos entre los médicos en formación y en profesionales de reciente graduación, pues debido a su baja morbilidad su diagnóstico puede pasar inadvertidos, así logramos también más y mejor eficiencia en la calidad integral del sistema de salud.

