










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Pediatría
versión On-line ISSN 1561-3119
Rev Cubana Pediatr v.70 n.2 Ciudad de la Habana abr.-jun. 1998
Neurofibromatosis tipo I con pubertad precoz. Presentación de un caso
Dra. Bárbara M. Esquejarosa Roque,2 Dr. Ricardo Hodelín Tablada,3 Dr. Víctor E. Salgueiro Medina,4 Dra. Mirtha Heredia Olivero5 y Dra. Katherine Cruz Egued6Resumen
La neurofibromatosis tipo I o neurofibromatosis de Von Reckinghausen es el trastorno neurocutáneo más frecuente. Se presenta en 1 de cada 3 000 personas y se hereda por vía autosómica dominante. Los criterios para su diagnóstico clínico fueron establecidos en 1988. Su asociación con la pubertad precoz, aunque se ha descrito, se considera poco frecuente. En nuestro trabajo se presenta un niño de 3 años con neurofibromatosis tipo I y pubertad precoz. Se destacan las manifestaciones clínicas y la importancia de los estudios imagenológicos para completar el diagnóstico.Descriptores DeCS: NEUROFIBROMATOSIS 1; PUBERTAD PRECOZ.
La neurofibromatosis tipo I (NF 1) o neurofibromatosis de Von Recking-hausen es el trastorno neurocutáneo más frecuente. Se presenta en 1 de cada 3 000 personas y se hereda por vía autosómica dominante.1,2 Los criterios para su diagnóstico clínico fueron establecidos en 1988.3 Desde el punto de vista clínico el trastorno se caracteriza por la presencia de manchas en la piel, con aspecto de café con leche, visibles generalmente desde el nacimiento.
Asimismo, pueden verse afectados el sistema nervioso central y el sistema nervioso periférico, el esqueleto, el sistema endocrino y los tejidos blandos. El gen transmisor se encuentra en el cromosoma 17q 11.2.2,4 La NF1 es para algunos autores la entidad que presenta complicaciones clínicas, radiológicas y patológicas más floridas y abundantes, que son las que hacen interesante este trastorno desde el punto de vista científico.2
En el actual trabajo presentamos el caso de un niño que consulta por pubertad precoz y que después del examen físico y los exámenes complementarios se llegó a la conclusión de que se trataba de una NF1. Se le consideró interesante si se tiene en cuenta la poca frecuencia con que se presenta esta asociación y se destaca la necesidad de un consejo genético a la familia y una atención médica multidisciplinaria.
REPORTE DEL CASO
Paciente masculino de 3 años de edad, nacido de parto eutócico intrahospitalario. Historia clínica 97-150184. Es el primer hijo de un matrimonio de jóvenes. Como antecedentes patológicos familiares que se deben destacar, el padre presenta NF1. Durante los primeros meses de la vida el niño mostró un desarrollo psicomotor normal para su edad. Después del primer año -según refiere los padres- comienzan a notarle crecimiento anormal de los genitales externos. Consulta al médico a los 3 años, y al examen físico presentó los siguientes aspectos (figs.1 y 2):
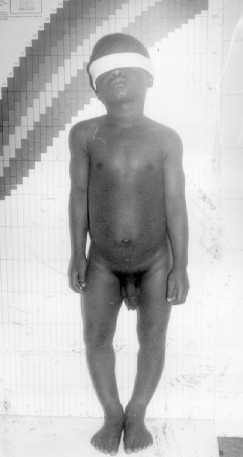
Fig. 1. Nótese la pubertad precoz y la deformidad en genus varo de miembros inferiores.
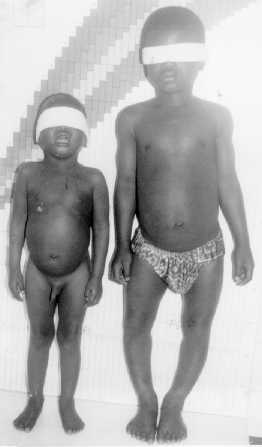
- Pubertad precoz.
- Escoliosis de convexidad derecha.
- Deformidades de ambos miembros inferiores en forma de genus varo.
- Puente nasal ancho.
- Hipertelorismo.
- Manchas café con leche de diferentes diámetros distribuidas en:
- Tono y trofismo globalmente aumentados.
- Voz grave.
- Talla elevada para su edad.
- Curva talla-edad: Más del 97 percentil.
- Curva peso-talla: Más del 97 percentil.
- Curva peso-edad: Más del 97 percentil.
- Radiografía de cráneo: No presencia de signos de hipertensión endocraneana. Implantación anómala de dientes en la arcada inferior.
- Radiografía de tibia y peroné: Afinamiento de la cortical del hueso peroné derecho (fig. 3).
- Radiografía de manos: Edad ósea compatible con un niño de 11 años.
- Ecografía: Testículos de diámetro de 4,5 cm, compatibles con un niño de 11 años.
- Tomografía axial computadorizada de cráneo: Ventrículos de tamaño normales. No hay lesión que ocupe espacio. No existen desplazamientos de las estructuras de línea media.
- Estudios hormonales: Alfafetoproteína, LH, FSH, prolactina, cortisol, testosterona libre, 17-OH-progesterona: normales.
- Somatotropina: Elevada.
- VN: 0-14, resultado 19,2 mm/L.
- Fondo de ojo. Pupilas de bordes bien definidos, vasos normales. No se aprecian alteraciones fondoscópicas.
- Radiografía de tórax anteroposterior y perfil: sin alteraciones.

El niño se valoró por un equipo multidisciplinario integrado por especialistas en pediatría, neurocirugía, oftalmología, ginecoobstetricia y ortopedia. Se les dio a los padres el consejo genético adecuado y se les orientó sobre la posibilidad del nacimiento de otros niños con la enfermedad. No pudimos realizarle otros estudios genéticos por las características del hospital donde trabajamos. En estos momentos se encuentra en fase preparatoria para efectuarse corrección de la deformidad de los miembros inferiores por parte de los ortopédicos.
COMENTARIOS
La NF1 entra dentro del grupo de las enfermedades llamadas facomatosis, término introducido en 1923 por Van der Hoeve5 y que proviene de "phakos", manchas más o menos elevadas de origen congénito, familiar y hereditario y "phakomas" o tumores extracutáneos, preferentemente retinianos. Estas enfermedades tienen en común 5 características fundamentales:6- Naturaleza displásica: Se debe a anomalías del desarrollo embrionario interesando principalmente las formaciones de origen ectodérmico -piel, sistema nervioso, retina-, así como los elementos vasculares de estos órganos.
- Tendencia blastomatosa: Poseen habitualmente un potencial de crecimiento excesivo con frecuente desarrollo de hamartomas y tumores benignos o malignos, a veces múltiples.
- Carácter heredofamiliar: La mayoría condicionados genéticamente, otras esporádicas y debidas a mutaciones, las menos de origen desconocido.
- Afectación multisistémica: En general hay varios órganos comprometidos.
- Heterogeneidad en su expresividad clínica: La semiología clínica varía desde formas frustradas con expresión muy localizada, hasta formas ricas en síntomas y signos.
Los criterios diagnósticos de la NF1 son:7
- Un mínimo de 6 manchas de color café con leche.
- Por lo menos 2 neurofibromas o un neuroma plexiforme.
- Manchas de salvado, pecas (freckling) axilares e inguinales.
- Glioma del nervio o del quiasma óptico.
- Por lo menos 2 hamartomas del iris.
- Lesiones óseas (adelgazamiento de la cortical de un hueso largo, pseudoartrosis).
- Familiar de primer grado (padre, hijo, hermano) que presentó un NF1.
En una serie de 100 casos publicados por Pou-Serradell7 la pubertad precoz estuvo presente conjuntamente con otras lesiones que alteran el sistema endocrino como el feocromacitoma y el adenoma hipofisario. El estudio de las alteraciones endocrinas y las dosificaciones hormonales en niños con pubertad precoz, ha tenido un gran desarrollo en los últimos años.9,10 Otra de las manifestaciones clínicas de NF1 presente en el caso que presentamos es la escoliosis, hallazgo frecuente en los casos de Pou-Serradell. Para este autor, la escoliosis constituye un signo muy importante, de tal forma que ante una escoliosis considerada idiopática han de buscarse signos de NF1, manchas café con leche y/o antecedentes familiares.7
Queremos destacar en el caso que presentamos, otras manifestaciones clínicas que no hemos hallado en la literatura médica revisada, como son la presencia de genus varo lo que le dificulta la bipedestación, voz grave, hipertelorismo y la implantación anómala de los dientes en la arcada inferior. Todos estos hallazgos clínicos y radiológicos entran dentro de la afectación multisistémica y la heterogeneidad en la expresividad clínica, que como señalamos anteriormente, caracterizan a este grupo de enfermedades.
Finalmente consideramos importante el consejo genético a la familia si se tiene en cuenta el carácter heredofamiliar de la enfermedad y la necesidad de que el niño sea tratado por un equipo de trabajo multidisciplinario, donde intervengan diferentes especialistas en dependencia de los órganos y sistemas afectados, que como hemos analizado pueden ser varios.
Summary
Neurofibromatosis type I ort Von Reckinghausen´s disease is the most common neurocutaneous disorder. It appears in one of every 3 000 persons and it is inherited by dominant autosomal route. The criteria for its diagnosis were established in 1988. Its association with preccocius puberty, although it has been described, is considered as rare. A 3-year-old child with neurofibromatosis type I and precocious puberty is presented in this paper. Emphasis is made on the clinical manifestation and on the importance of the image studies to complete the diagnosis.Subject hedings: NEUROFIBROMATOSIS I; PUBERTY, PRECOCIOUS.
REFERENCIAS BIBLIOGRÁFICAS
- Ricardi VM. Von Rechminghausen neurofibromatosis. N Engl J Med 1981;305:1617-27.
- Pascual-Castroviejo I. Neurofibromatosis tipo I (NF1): peculiaridades y complicaciones. Rev Neurol (Barc) 1996; 24 (133):1051-5.
- National Institute of Health Consensus Development Conference; neurofibromatosis conference statement. Arch Neurol 1988; 45:575-8.
- Viskochil D, Buchberg AM, Xu G. Deletion and a translocation interrupt a cloned gene at the neurofibromatosis type 1 locus. Cell 1990;68:187-92.
- Van der Hoeve J. Eye symptoms in tuberous sclerosis of the brain and Recklinghausen disease. Trans Ophthamol Soc UK 1923;534-41.
- Nieto M. Facomatosis. Criterios diagnósticos y protocolos de seguimiento: introducción. Rev Neurol (Barc) 1996; 24(133): 1949-50.
- Pou-Serradell A. Evolución natural de las facomatosis en la edad adulta. Rev Neurol (Barc) 1996;24(133):1085-127.
- Habiby R, Silverman B, Listernick R, Charrow J. Precocious puberty in children with neurofibromatosis type 1. J Pediat 1995;126(3):364-7.
- Gupta R, Ammini AC. Precocious puberty with pituitary gland hyperplasia: two cases in one f amily. Pediatr Radiol 1996;26(6):418-20.
- Yano K, Kohn LD, Seji M, Kataora N, Okuno A, Cutter GB Jr. A case of male limited precocious puberty caused by a point mutation in the second transmembrane doman of the luteinizing hormona chriogonadotropin receptor gene. Biochem Biophys Res Communic 1996;220(3):1036-42.
Dra. Bárbara M. Esquejarosa Roque. Calle G, número 32, entre 4ta y Norte, reparto Celso Magaroto. Pinar del Río, Cuba.
1 Trabajo presentado en la X Jornada de Salud y I Congreso Luso- Mozambicano de Infectología. Maputo, Mozambique, del 8 al 12 de septiembre de 1997.
2 Especialista de I Grado en Pediatría.
3 Especialista de I Grado en Neurocirugía.
4 Especialista de I Grado en Ginecoobstetricia
5 Especialista de I Grado en Oftalmologìa
6 Residente de Medicina General integral
4 Especialista de I Grado en Ginecoobstetricia.
5 Especialista de I Grado en Oftalmología.
6 Residente de Medicina General Integral.

