










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Pediatría
versión On-line ISSN 1561-3119
Rev Cubana Pediatr v.71 n.3 Ciudad de la Habana jul.-set. 1999
Reporte de casos
Centro Nacional de Genética Médica. Instituto Superior de Ciencias Médicas de La HabanaMixoploidía Diploide - Tetraploide: Primer Reporte en Nuestro Medio
Lic. Héctor Pimentel Benítez,1 Dra. Aracely Lantigua Curz2 y Lic. Olga Quiñones Maza3RESUMEN
Se presenta un caso de mixoploidía diploide-tetraploide, con ligeros signos clínicos pertenecientes a esta condición de la poliploidía en humanos. Estudios citogenéticos a partir de cultivos de sangre periférica y fibroblastos de piel permitieron establecer la fórmula cromosómica 46,XX/92XXXX en la paciente. Se realiza correlación fenotipocariotipo, así como su presentación clínica. Este estudio de casos ayuda a enriquecer el conocimiento sobre esta condición, contribuye a su mejor diagnóstico y a dilucidar la etiología de síndromes malformativos inespecíficos.Descriptores DeCS: POLIPLOIDIA; MOSAICISMO; RETARDO MENTAL.
Las poliploidías como aberración cromosómica fue estudiada como una condición concerniente únicamente al reino vegetal; sin embargo, en los humanos eran consideradas como condición letal, aunque células somáticas como los hepatocitos son normalmente poliploides.
Las poliploidías más frecuentemente detectadas en nuestra especie son las triploidías y tetraploidías completas, además mixoploides diploide/triploide (2n/3n) y diploide/tetraploide (2n/4n), en estas últimas, coexiste una población de células normales diploides (2n), con otra que tiene 3 o más múltiplos haploide de cromosomas, por ejemplo: triploide (3n) o tetraploide (4n).
Autores como Kohn (1967), Kelly y Ray (1974), Redd (1977), Veenema (1982), Quiroz (1985), Willson (1988), Moris y Lustgarten (1989), Urioste (1990) y Willich (1991), han reportado casos de mosaicos 2n/4n, los que completan la cifra aproximada de 30 reportes hasta el año 1994,1-11 quienes resaltan sus hallazgos clínicos, que enriquecen el conocimiento del fenotipo mixoploide 2n4n y tratan de explicar los mecanismos de formación de esta condición.
Basados en esta información y con el objetivo de contribuir al diagnóstico de síndromes malformativos inespecíficos, hacemos la presentación de una paciente que muestra signos clínicos ligeros, pertenecientes al mosaicismo 2n/4n, el que fue confirmado citogenéticamente en estudios a partir de sangre periférica y fibroblastos de piel.
REPORTE DEL CASO
Paciente femenina, mestiza, de 18 años de edad, nacida a las 42 semanas de gestación, después de un embarazo sin complicaciones, de una primigesta saludable.Edad materna y paterna al nacimiento de 23 y 28 años respectivamente.
Parto eutócico, peso al nacer 2,50 kg (perteneciente al tercer percentil), talla 46,1 cm, circunferencia cefálica de 36 cm.
EXAMEN FÍSICO
En éste se observaron cabezas con fontanelas normales, frente prominente, ligera asimetría facial a expensas del lado derecho, el que es más voluminoso que el izquierdo; nariz prominente. Hemicuerpo derecho más voluminoso que el izquierdo. Genitales normales.Extremidades: Miembro superior derecho más voluminoso que el izquierdo y ligera desproporción en longitud. En las manos se observan pliegues de flexión anormales y clinodactilia del 5to. dedo de la mano derecha. Se aprecian extensión limitada de los codos de ambos brazos, cadera derecha más alta que la izquierda y miembro inferior derecho más largo y voluminoso que el izquierdo. Mantiene buen tono muscular.
En la piel mostró zonas hiperpigmentadas, una periumbilical derecha y otra en la escápula izquierda. Presenta retraso mental ligero.
ESTUDIOS CITOGENÉTICOS
Éstos se realizaron en los tejidos supuestamente afectados por esta condición y los más comúnmente utilizados, sangre periférica y fibroblastos de piel.Sangre periférica: Se obtuvo la muestra con jeringuillas previamente heparinizadas.
La siembra se realizó según el microcultivo, utilizando medio RPMI 1640. Los cromosomas se cosecharon según el procedimiento seguido por Verma y Babú en 1995 (protocolo 2,5).12 Se realizó bandeo GTG y se analizaron 200 metafases.
Fibroblastos de piel: Con este fin, se realizó biopsia de piel a la paciente según técnica de rutina empleada por el Servicio de Dermatología del Hospital Pediátrico Docente "Juan Manuel Márquez", de Ciudad de La Habana, y se obtuvo muestra de tejido de una zona del cuerpo con crecimiento hipertrófico e hiper o hipopigmentado.
Las células crecieron en medio de cultivo Eagle MEM (Gibco), suplementado con 10 % de suero fetal bovino (Gibco) y antibiótico combinado de penicilina-estreptomicina.
El bandeo cromosómico que se utilizó fue GTG y se analizaron 200 metafases para cada hemicuerpo.
En el estudio de ambos tejidos, se incluyó un caso control, el que se cultivó y procesó en iguales condiciones que el de la paciente, con el objetivo de valorar la aparición de poliploidías in vitro, por ser éstos a mediano y largo plazos.
En todos los procesos de cosechas cromosómicas se utilizó Karyomax Colcemid Solution, que se certifica como un agente antimitótico efectivo en cultivos de tejidos, que tiene actividd similar a la colchicina.13
Se tuvo en cuenta la composición del complemento cromosómico (número de cromosomas) en todas las metafases poliploides detectadas, para establecer el diagnóstico.
RESULTADOS
En la mixoploidía 2n/4n el complemento cromosómico de la línea anormal es de 92 cromosomas; en nuestra paciente éste se detectó en 3 células de las 200 analizadas, a partir de linfocitos de sangre periférica, lo que representa el 1,5 % de células 4n y el 98,5 % de células 2n.Estos hallazgos cromosómicos no permiten afirmar el diagnóstico de la condición mosaico, ni tampoco sugieren la no aparición de ella en otros tejidos, razón por la cual se practicó biopsia de piel, como otro tejido de relativo fácil acceso.
Los resultados de los estudios cromosómicos a partir de cultivos de fibroblastos de piel, se resumen en la figura, donde la línea celular aberrante sólo se observó en las preparaciones obtenidas de aquellos cultivos pertenecientes al tejido macrosómico y que en particular mostraba un pequeño parche hiperpigmentado.
El establecimiento de la fórmula cromosómica 46,XXI 92, XXXX, corrobora la ya establecida en el análisis realizado en sangre periférica. La línea celular 2n se determinó en el 86,5 % de las células analizadas del hemicuerpo afectado por la macrosomía, mientras que 27 metafases portaron el complemento tetraploide, lo que representó el 13,5 % del total de células analizadas en este hemicuerpo.
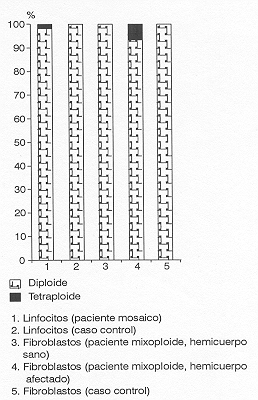
El otro hemicuerpo mostró el 100 % de células 2n.
Los resultados del estudio cromosómico en el caso control, reflejaron complemento cromosómico normal en todas las células analizadas, lo que indicó que los cultivos crecieron en óptimas condiciones, donde no hubo inducción, ni formación de poliploidías in vitro.
La identificación de signos clínicos pertenecientes a los descritos en el mosaicismo 2n/4n, de nuestra paciente, entre ellos la asimetría, pigmentación irregular de la piel, retraso mental ligero y CIUR, permitieron establecer cierta correlación fenotipo-cariotipo, y resaltar que sus signos clínicos no son severos.
El hecho de que en la paciente con sospecha clínica de mixoploide exista el diagnóstico de un mixoploide 2n/4n, nos permite plantear que es necesaria la inclusión de este tipo de estudio, como medio de cooperación para el diagnóstico etiológico de casos con retraso mental y asimetrías corporales en nuestro medio.
El examen de 2 tipos de tejidos, los comúnmente cariotipados, permitieron ofrecer la demostración y evidencia de la poliploidía (4n) en esta paciente, lo que unido a los hallazgos clínicos permitió establecer el diagnóstico de mixoploide 2n/4n, en la paciente estudiada, descrito por primera vez en nuestro medio.
La determinación del origen parental del set cromosómico extra, no se evidenció citogenéticamente en este caso.
DISCUSIÓN
En la literatura médica consultada14 se reporta que en menos del 5 % de las metafases analizadas de la población linfocitaria, proveniente de individuos con sospechas clínicas de portar alguna de las condiciones de mixoploidía en humanos se observan, lo que coincide con nuestros resultados; además, en linfocitos, las líneas celulares trisómicas, tetrasómicas y poliploides, con el tiempo tienden a cambios, los que están dirigidos hacia la eliminación de éstas,15 hecho que favorece a la baja frecuencia de aparición de estas células; otros autores plantean que el hallazgo de células poliploides en tan bajo porcentaje en los linfocitos, se deba a que éstas se encuentran en los complejos procesos de hematopoyesis, en franca desventaja selectiva, respecto a las células 2n, e incluso, en muchos casos se pueden perder antes de comenzar a circular.8Si comparamos los signos clínicos de la paciente con los mostrados por Edwards, y otros, 1994,16 éstos se corresponden con el bajo tanto por ciento de células tetraploides caracterizadas en los estudios cromosómicos realizados, lo que demuestra que el fenotipo relativamente normal, es cualitativo y cuantitativamente diferente a otros informados. Rojanasakut, y otros, 1985 y Scarbrough, y otros, 1984, demostraron en 2 casos reportados, que, el bajo porcentaje de mosaicismo 2n/4n está asociado con hallazgos clínicos normales o ligeramente anormales,17,18 con lo que se corresponde nuestro estudio.
Se sugiere generalmente que la condición de mixoploide, se origina por cambios ocurridos en la primera división del cigoto o muy cercanos a ésta, y que otras hipótesis plantean su origen a partir de la fusión de gametos diploides o menos probables que ocurran por procesos de partenogénesis.8
En conclusión podemos decir:
- A partir de cultivos de sangre periférica y fibroblastos de piel, se efectuó el primer diagnóstico de un mixoploide 2n/4n en nuestro medio.
- El uso de un caso control en los cultivos de tejidos realizados demostró ser lo suficientemente fiel, para fundamental que las condiciones de cultivo no favorecieron la producción de poliploidías in vitro.
- El diagnóstico de mixoploides debe tenerse en cuenta frente a síndromes dismórficos asimétricos con gradaciones de retraso mental.
- La ignorancia relativa existente aún en la expresión fenotípica de estos defectos genómicos en el humano, nos permiten sugerir la continuidad y sistematicidad de investigaciones que como ésta, están a nuestro alcance.
SUMMARY
A case of diploid-tetraploid mixoploidy with mild clinical signs correspondoing to this condition of polyploidy in humans is presented. Cytogenetic studies based on cultures of peripheral blood and fibroblasts of skin allowed to obtain the 46,XX/92XXXX chromosomic formula in the patient. The phenotype-kariotype correlation was stablished and its clinical presentation was made. This study of cases helps to improve the knowledge about this condition, and it also contributes to have a better diagnosis and to dilucidate the etiology of unspecific syndromes of malformation.Subject Headings: POLYPLOIDY; MOSAICISM; MENTAL RETARDATION.
REFERENCIAS BIBLIOGRÁFICAS
- Kohn G, Mayall DE. Tetraploid-diploid mosaicism in surviving infant. Pediatr Res 1967;1:461-9.
- Kelly TE, Ray JE. Mosaic tetraploidy in two years old femele. Clin Genet 1974;6:221-4.
- Redd CM, Sing DE. Diploid-tetraploid mosaicism in a malformed boy. Clin Genet 1985;27:183-6.
- Veenema H. Mosaic tetraploidy in a male neonate. Clin Genet 1982;22:295-8.
- Quiroz E, Orosco A. Diploid-tetraploid mosaicism in a malformed boy. Clin Genet 1985;27:183-6.
- Willson GN. MCA/MR syndrome in a female with tetraploidy mosaicism: review of the human poliploidy phenotype. Am J Med Genet 1988;30:753-61.
- Wittwer BB. Information about diploid-tetraploid mosaicism in a six year old male. Letter to editor. Clin Genet 1985;18:567-8.
- Augthon DJ. Diploid-tetraploid mosaicism in a live birth infant demostrable only in the bone marrow: case report and literature review. Clin Genet 1988;33:299-307.
- Morris C, Lustgarden M. Metaphyseal lesions in mosaic tetraploidy. Proc Greenwood Genet 1989;8:220.
- Urioste M. Diploid-tetraploid mosaicicism a still birth infant with prune belly anomaly. An Genet 1990;33:48-51.
- Willich B, Henn W. Mosaic tetraploidy in a live birth infant with features of the Di George anomaly. Clin genet 1991;40:353.
- Verma RS, Babu A. Human chromosomes. Principles and techniques 2nd New York: Pergamon Press, 1995:9-29.
- Catalogue for cell-culture. New York: Life Technologies; 1997:92-3.
- Wolfsberg EA, Pob CA. Monozygotic twin girls with diploid-triploid chromosome mosaicism and cutaneous pigmentary displase. Clin Genet 1991;35:370-5.
- Kupke KG, Berry PK, Muller U. Diploid-triploid mixoploidy: case report and use of cytogenetic heteromorphism and DNTR polymorphism to determine parenteral origin and nature of chromosome loss. Am J Human Genet 1992;4:51-6.
- Edwards MJ. Mixoploidy in humans: two surviving case of diploid-tetraploid mixoploidy. Am J Med Genet 1994;52:324-30.
- Rojanasakut A. Tetraploid in two sister with polycystics ovary syndrome. Clin Genet 1985;27:167-74.
- Scarbrought PR. Tetraploidy: a report of three live birth infants. Am J Med Genet 1984;19:29-37.
Dr. Héctor Pimentel Benítez. Calle 2, número 6, entre A y Final, Reparto Las Mercedes, Camagüey, 71200, Cuba.
2 Doctora en Ciencias Médicas. Departamento de Genética Clínica. Centro Nacional de Genética Médica.
3 Licenciada en Ciencias Biológicas. Investigadora Agregada. Departamento de Citogenética. Centro Nacional de Genética Médica.

