










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Pediatría
versión On-line ISSN 1561-3119
Rev Cubana Pediatr v.73 n.3 Ciudad de la Habana jul.-set. 2001
Artículos de revisión
Hospital Pediátrico Universitario William Soler
Epilepsias mioclónicas en el niño y el adolescente
Dra. Albia J. Pozo Alonso,1 Dr. Desiderio Pozo Lauzán2 y Dr. Desi Pozo Alonso3
Resumen
Las mioclonías epilépticas son contracciones musculares rápidas originadas por una descarga que proviene del sistema nervioso central y que se identifican por la correlación del electroencefalograma con la sacudida muscular. Las epilepsias mioclónicas son un grupo de síndromes epilépticos que evolucionan exclusiva o preferentemente con crisis mioclónicas. De acuerdo con su causa pueden clasificarse en idiopáticas, sintomáticas y criptogénicas. Según su evolución pueden ser benignas, graves y progresivas. Fue propósito de este trabajo mostrar las características más importantes de algunos de los principales síndromes epilépticos mioclónicos y enfatizar en las manifestaciones clínicas, hallazgos electroencefalográficos y en el tratamiento empleado.
DeCS: EPILEPSIAS MIOCLONICAS/diagnóstico; EPILEPSIAS MIOCLONICAS/clasificación; epilepsiaS mioclonicas/quimioterapia; ELECTROENCEFALOGRAFIA; ACIDO VALPROICO/uso terapéutico; CLONAZEPAM/uso terapéutico; BENZODIAZEPINAS/uso terapéutico; niño; ADOLESCENCIA.
Las mioclonías son sacudidas musculares súbitas, involuntarias, cortas, que se originan en el sistema nervioso central.1
Desde el punto de vista semiológico2 las mioclonías se pueden clasificar según su distribución topográfica en generalizadas, segmentarias, focales y multifocales. De acuerdo con su patrón temporal, en rítmicas y arrítmicas.
En relación con su forma de inducción, en espontáneas, de acción (por actividad muscular) y reflejas (provocadas por estímulos sensoriales o somestésicos).
En función de su fisiopatología en corticales, subcorticales, corticosubcorticales y espinales. Teniendo en cuenta su frecuencia3 en aisladas o repetidas.
Las mioclonías epilépticas son contracciones musculares rápidas originadas por una descarga que proviene del sistema nervioso central y que se identifican por la correlación del electroencefalograma con la sacudida muscular.
El electroencefalograma (EEG) ictal muestra puntas múltiples, rápidas, de alto voltaje, seguidas de ondas lentas, con un patrón interictal de punta, polipunta, punta-onda lenta o polipunta-onda lenta.4 Deben diferenciarse de espasmos epilépticos y ataques tónicos, de mioclonías no epilépticas, así como de tics, tremor y corea.5
Las epilepsias mioclónicas son un grupo de síndromes que evolucionan exclusiva o preferentemente con crisis mioclónicas.6
De acuerdo con su origen pueden clasificarse en idiopáticas, sintomáticas y criptogénicas.
Las epilepsias mioclónicas idiopáticas están relacionadas con la edad de aparición y la existencia de una fuerte predisposición genética y tienen una evolución favorable; en las sintomáticas, se incluyen formas progresivas asociadas a procesos metabólicos y degenerativos y otras que son secundarias a encefalopatías no progresivas, generalmente de origen prenatal o perinatal y en las criptogénicas la causa no es conocida, aunque se presume que sean sintomáticas.6,7
En la figura 1 puede observarse un EEG correspondiente a un paciente de 8 años de edad que muestra una epilepsia mioclónica sintomática.
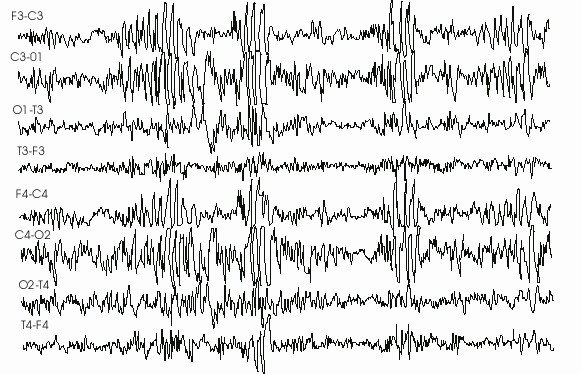
Fig. 1. Se observan descargas de puntas y polipuntas-ondas generalizadas que se agrupan en oleadas.
Los principales síndromes epilépticos mioclónicos son los siguientes:
- Encefalopatía mioclónica temprana.
- Epilepsia mioclónica benigna de la infancia.
- Epilepsia mioclónica benigna refleja de la infancia.
- Epilepsia mioclónica familiar de la infancia.
- Epilepsia mioclónica severa de la infancia.
- Epilepsia mioclónica en encefalopatías no progresivas.
- Epilepsia mioclónica-astática.
- Epilepsia con ausencias mioclónicas.
- Mioclonías palpebrales con ausencias.
- Mioclonías periorales con ausencias.
- Epilepsia mioclónica juvenil.
- Epilepsias mioclónicas progresivas.
Encefalopatía mioclónica temprana
Se incluye en el grupo de las epilepsias y síndromes epilépticos generalizados sintomáticos sin causa específica.7 Las manifestaciones clínicas comienzan en la mayoría de los pacientes en el período neonatal y se caracteriza fundamentalmente por la presencia de:4,8
- Mioclonías erráticas fragmentarias o parciales.
- Mioclonías masivas.
- Crisis parciales simples.
- Espasmos infantiles de tipo tónico.
Es una forma muy rara de epilepsia. La incidencia familiar es frecuente y presenta múltiples causas. Es posible la ocurrencia de herencia recesiva en algunos casos, pero el patrón genético no es constante. Puede deberse a diversos errores metabólicos congénitos y también a malformaciones cerebrales.4
Las mioclonías segmentarias y erráticas constituyen la manifestación crítica más precoz y pueden presentarse desde las primeras horas de vida. Las sacudidas afectan la cara o los miembros y se limitan a un territorio como un dedo o afectan todo un miembro. En la mayoría de los pacientes las mioclonías se repiten frecuentemente o son casi continuas y pasan de un territorio al otro, de una forma asincrónica y anárquica.
Las mioclonías masivas pueden aparecer precozmente y alternar con mioclonías erráticas. Las crisis parciales aparecen poco tiempo después que las mioclonías erráticas y se manifiestan por una desviación de los ojos con movimientos clónicos o sin éstos y también se caracterizan por manifestaciones autonómicas como una apnea, rubicundez facial, entre otras.qw2< Los espasmos infantiles tónicos se presentan generalmente alrededor de los 3 a 4 meses de edad.4,8
El EEG muestra una actividad de base anormal. Durante el sueño y la vigilia se observan complejos de puntas, ondas agudas y ondas lentas irregulares y arrítmicas de una duración de 1 a 5s separados por períodos de aplanamiento del trazado de 3 a 10 s de duración. Después de los 3 a 5 meses de edad este patrón puede cambiar a hipsarritmia atípica o a paroxismos multifocales.8
La evolución es desfavorable. Todos los niños presentan anomalías neurológicas graves y la mayoría de ellos fallecen antes del año de edad. Estos pacientes no responden adecuadamente a los medicamentos antiepilépticos como la hormona adrenocorticotropa (ACTH), corticoides o piridoxina.8
Epilepsia mioclónica benigna de la infancia
De acuerdo con la Clasificación Internacional de las Epilepsias, Síndromes Epilépticos y Trastornos Paroxísticos relacionados, se incluye dentro de los síndromes epilépticos generalizados idiopáticos.7 Fue descrita en 19819 y representa el 7 % de las epilepsias mioclónicas.10 Es más frecuente en los varones y la edad de comienzo de las crisis se sitúa entre los 6 meses y los 2 años. Existen antecedentes familiares de epilepsia o convulsiones en el 25-30 % de los casos. El desarrollo psicomotor y el examen neurológico son siempre normales, aunque los niños que no reciben un tratamiento precoz pueden tener dificultades escolares o trastornos de la personalidad.3,11
Clínicamente se manifiesta por mioclonías generalizadas que afectan al eje corporal y a los miembros con caída de la cabeza sobre el tronco y movimiento de elevación-abducción de miembros superiores y flexión de los inferiores con desequilibrio o caída sin pérdida de conciencia. Las crisis son breves, habitualmente de 1 a 3 s de duración, aunque pueden prolongarse hasta 8-10 s y se presentan aisladas.3,10,11 Por lo general son diurnas, sin relación horaria y desaparecen durante el sueño profundo. La fotoestimulación puede desencadenarlas. Algunos pacientes presentan crisis tónico-clónicas generalizadas en la adolescencia.3,4 El EEG interictal muestra en vigilia una actividad de base normal, mientras que en el EEG ictal se observan descargas breves de punta-onda o polipunta-onda generalizadas y rápidas que se incrementan con la fotoestimulación y la somnolencia. El EEG de sueño durante las primeras fases de sueño lento muestra descargas de punta-onda o polipunta-onda generalizadas con traducción clínica o sin ésta.3,4 El medicamento de elección es el valproato de sodio con el cual se logra el control de las crisis.10-12
Epilepsia mioclónica benigna refleja de la infancia
Se considera un nuevo síndrome epiléptico generalizado idiopático que presenta manifestaciones similares a las de la epilepsia mioclónica benigna de la infancia;13 sin embargo, las mioclonías no aparecen espontáneamente, pues son provocadas por la estimulación auditiva o táctil.3,11 Las mioclonías reflejas se inician entre los 6 y 21 meses de edad en niños de uno y otro sexos que presentan un desarrollo psicomotor normal. Se localizan en los miembros superiores o la cabeza y son desencadenadas por los estímulos táctiles o sonoros. En 2/3 de los casos aparecen crisis espontáneas facilitadas por la somnolencia y el sueño ligero, unos meses después del inicio de las crisis reflejas.3
El EEG interictal es normal en vigilia. El EEG ictal muestra descargas muy breves de puntas y polipunta-onda bilaterales a 3 ciclos/s, tanto en vigilia como en el sueño.3 La evolución es favorable con desaparición de las mioclonías reflejas, espontáneamente o después de la administración del valproato de sodio.3,11
Epilepsia mioclónica severa de la infancia
Pertenece al grupo de las epilepsias y síndromes indeterminados con crisis focales y generalizadas.7 Constituye el 29 % de las epilepsias mioclónicas y se observa en 1 de cada 40 000 niños.14 Se presenta durante el primer año de vida, como promedio entre los 5 a 6 meses de edad. Predomina el sexo masculino.
Existen antecedentes familiares de epilepsia o convulsiones en el 25 % de los pacientes.15 Se inicia con crisis febriles clónicas generalizadas o unilaterales, de larga duración y recurrentes. A medida que aumentan las recurrencias, la fiebre es cada vez más discreta y a partir de los 18 a 24 meses aparecen crisis afebriles.3
Hacia la mitad del segundo año aparecen las crisis mioclónicas generalizadas, que son aisladas, frecuentes y a veces continuas. Los ataques parciales se presentan algo más tarde o al mismo tiempo que las crisis mioclónicas y se caracterizan por manifestaciones autonómicas, pérdida del tono o automatismos. Pueden generalizarse en forma de crisis clónicas o hemiclónicas. También aparecen estados de mal no convulsivo con mioclonías palpebrales parcelares3,11 y ausencia atípicas.
El EEG interictal es normal durante los primeros meses de evolución. Sin embargo, entre los 18 a 24 meses se aprecia fotosensibilidad y posteriormente descargas de punta-onda y polipunta-onda gene-ralizadas, aisladas o en salvas breves. En ocasiones se observan anomalías focales o multifocales.3
La evolución es desfavorable, y aparecen a partir de los 2 años retraso mental cada vez más marcado y posteriormente torpeza motora y ataxia.3,11
En esta entidad se han encontrado diversas alteraciones inmunológicas, y son las más frecuentes las relacionadas con las inmunoglobulinas séricas. Es posible que en esta epilepsia intervengan factores genéticos y factores ambientales y que las alteraciones inmunológicas encontradas faciliten reacciones de tipo hiperérgico en determinadas circunstancias.16
Habitualmente las crisis son refractarias al tratamiento. Se han obtenido mejorías parciales con valproato de sodio y benzodiazepinas.3 Con el empleo de inmunoglobulinas por vía endovenosa se ha logrado reducir las crisis en el 30 % de los casos tratados.17 Se han obtenido buenos resultados al asociar el valproato de sodio al clobazam.18 Otras alternativas terapéuticas la constituye la asociación de topiramato con valproato de sodio y muestan eficacia en algunos estudios19 y también la asociación de stiripentol con valproato de sodio y clobazam.20
Epilepsia mioclónica en encefalopatías no progresivas
Las manifestaciones clínicas se inician durante el primer año de vida. Se caracteriza por la repetición de estados de mal mioclónicos de larga duración con un severo déficit motor e intelectual. Presentan además ausencias breves pero frecuentes, acompañadas de mioclonías palpebrales o de sacudidas de los ojos y de los músculos distales.21 Se debe a causas perinatales o genéticas (anomalías cromosómicas o múltiples anomalías congénitas).22
El EEG interictal durante la vigilia muestra una actividad de base lenta asociada a anomalías focales o multifocales que predominan en las regiones parieto-occipitales. El EEG ictal se caracteriza por la presencia de breves descargas de punta-onda lenta difusas.21
La evolución es desfavorable. Las crisis son refractarias al tratamiento. La asociación de ácido valproico con etosuximida disminuye la frecuencia de las crisis.21
Epilepsia mioclónica juvenil
Constituye un síndrome epiléptico generalizado idiopático.7 Es la epilepsia mioclónica más frecuente. Representa aproximadamente el 10 % de todas las epilepsias.22 Con frecuencia existen antecedentes familiares de epilepsia.11,22 El locus para el gen de esta epilepsia se encuentra en el brazo corto del cromosoma 6(6p21.3)23 y también se ha identificado en el brazo largo del cromosoma 15 (15q14),24 lo que sugiere heterogeneidad genética. Las manifestaciones clínicas se inician habitualmente entre los 12 a 18 años, y afectan uno y otro sexos por igual. Se caracteriza por la presencia de crisis mioclónicas, tónico-clónicas generalizadas y ausencias típicas.
Las crisis mioclónicas se manifiestan como sacudidas musculares breves, aisladas o agrupadas que afectan principalmente los miembros superiores de forma bilateral. Si se presentan en los miembros inferiores pueden provocar la caída del paciente. No se acompañan de pérdida de la conciencia. Generalmente se manifiestan por las mañanas, casi siempre al despertar o unos minutos más tarde.4,11,22 En el 42 % de los pacientes se han reportado mioclonías asimétricas o unilaterales.25 Las crisis tónico-clónicas generalizadas presentes en el 90 % de los casos ocurren habitualmente al despertar.22 El examen neurológico y el intelecto son normales en estos pacientes. Como factores precipitantes de las crisis en este tipo de epilepsia se señala la privación de sueño, el estrés y el consumo de alcohol.25 También se reporta la fotoestimulación.22
El EEG de vigilia puede ser normal; sin embargo, con frecuencia se observan descargas generalizadas de punta-onda y polipunta-onda.26 Es frecuente la fotosensibilidad.27 En algunos pacientes pueden observarse paroxismos con predominio focal en uno y otro hemisferio que pueden conducir a errores diagnósticos de epilepsia parcial. El EEG de sueño muestra en todos los pacientes las anomalías paroxísticas mencionadas.26
Estudios neuropsicológicos sugieren disfunciones en el lóbulo frontal en pacientes con este tipo de epilepsia. Se ha demostrado por estudios realizados mediante espectroscopia por resonancia magnética, que en estos pacientes existen concentraciones reducidas de N-acetil aspartato en la región prefrontal.28
El medicamento de elección es el ácido valproico.12,22,29-31 También ha demostrado ser eficaz la lamotrigina empleada en monoterapia o asociada al ácido valproico.12,22 Las benzodiazepinas (clonazepam o clobazam) representan otra alternativa terapéutica.29
El pronóstico de esta entidad es bueno,11 aunque se ha referido la reaparición de las crisis con la suspensión del tratamiento.22,29
Epilepsias mioclónicas progresivas
Se incluyen dentro de las epilepsias generalizadas sintomáticas.7
Constituyen aproximadamente el 1 % de las epilepsias del niño y del adolescente.32 Se trata de trastornos genéticos que se caracterizan por presentar diferentes tipos de crisis epilépticas y predominan las mioclonías, deterioro intelectual y ataxia.33 también se refieren trastornos visuales progresivos.11,34
Las crisis que más frecuentemente se asocian a las crisis mioclónicas son las tónico-clónicas generalizadas, las parciales y también se ha observado, aunque con menor frecuencia, la ocurrencia de crisis de ausencia.34
Las principales epilepsias mioclónicas progresivas son las siguientes:
- Enfermedad de Unverricht-Lundborg.
- Epilepsia mioclónica progresiva tipo Lafora.
- Ceroidolipofuscinosis neuronal.
- Sialidosis.
- Citopatías mitocondriales.
El electroencefalograma es importante para el seguimiento, pronóstico y diagnóstico diferencial de las epilepsias mioclónicas progresivas. Durante la fase de progresión de la enfermedad de Unverricht-Lundborg la actividad de base no se modifica y la mejoría de las características paroxísticas bajo tratamiento con medicamentos antiepilépticos puede contribuir a sospechar de otras epilepsias mioclónicas progresivas como la enfermedad de Lafora, la ceroidolipo-fuscinosis y la sialidosis.35
Los medicamentos de elección son el valproato de sodio, el clonazepam y el piracetam. También se han utilizado los antioxidantes.33 La zonisamida asociada al clonazepam y al valproato se considera útil, aunque pueden ocurrir recurrencias.12 Estas epilepsias por lo general son refractarias al tratamiento.
Summary
The mioclonic epilepsies are rapid muscular contractions originated by a discharge that comes from the central nervous system and that are identified by the correlation of the EEG with the muscular jerk. The mioclonic epilepsies are a group of epileptic syndromes that evolve exclusively or preferably with myoclonic crises. They may be classified as idiopathic, symptomatic or cryptogenic according to their cause. They may be benign, severe or progressive depending on their evolution. The aim of this paper was to show the most important characteristics of some of the main mioclonic epileptic syndromes and to make emphasis on the clinical manifestations, EEG findings and the treatment used.
Subject headings: EPILEPSIES, MIOCLONIC/diagnosis; EPILEPSIES, MIOCLONIC/classification; EPILEPSIES, MIOCLONIC/drug therapy; ELECTRO-ENCEPHALOGRAPHY; VALPROIC ACID/therapeutic use; BENZODIAZEPINES/therapeutic use; CHILD; ADOLESCENCE.
Referencias bibliográficas
- Obeso JA, Artieda J, Marsden CD. Different clinical presentations of myoclonus. En: Jankovic J, Tolosa E, eds. Parkinsons disease and movement disorders. Baltimore: Urban and Schwarzenberg, 1988:263-74.
- Artieda J. Catalogación neurofisiológica de las mioclonías. Rev Neurol 1999;28:272-7.
- Nieto-Barrera M. Mioclonías y epilepsias mioclónicas en la infancia. Rev Neurol 1999;28:278-84.
- Medina-Malo C, Obando MT. Epilepsias mioclónicas en Pediatría. Rev Neurol 1999;28:407-16.
- Dulac O, Plouin P, Shewmon A. Myoclonus and epilepsy in childhood: 1996 Royaumont meeting. Epilepsy Res 1998;30:91-106.
- Aicardi J. Diseases of the nervous system in childhood. Clinics in developmental medicine. En: Lavenham S, ed. Epilepsy and other seizure disorders. London: Mac Keith Press, 1992:909-1000.
- Commission on Classification and Terminology of the International League Against Epilepsy. Proposal for revised classification of epilepsies and epileptic syndromes. Epilepsia 1989;30:389-99.
- Aicardi J. Encéphalophathie myoclonique précoce (encéphalopathie myoclonique néonatale). En: Roger J, Bureau M, Dravet C, Dreifuss FE, Perret A, Wolf P, eds. Les syndromes épileptiques de lenfant et de ladolescent. 2e éd. London: John Libbey, 1992:13-23.
- Dravet C, Bureau M. Lépilepsie myoclonique bénigne du nourrison. Rev EEG Neuropsysiol 1981;11:438-44.
- Dravet C, Bureau M, Roger J. Lépilepsie myoclonique bénigne du nourrison. En: Roger J, Bureau M, Dravet C, Dreifuss FE, Perret A, Wolf P, eds. Les syndromes épileptiques de l´enfant et de l´adolescent. 2e éd. London: John Libbey, 1992:67-74.
- Palencia R. Epilepsias mioclónicas en la infancia. Rev Neurol 2000;30:S15-S24.
- Wallace SJ. Myoclonus and epilepsy in childhood: a review of treatment with valproate, ethosuximide, lamotrigine and zonisamide. Epilepsy Res 1998;29:147-54.
- Ricci S, Cusmai R, Fusco L, Vigevano F. Reflex myoclonic epilepsy in infancy: a new age-dependent idiopathic epileptic syndrome related to startle reaction. Epilepsia 1995;36:342-8.
- Hurts DL. Epidemiology of severe myoclonic epilepsy of infancy. Epilepsia 1990;31:397-400.
- Dulac O, Arthuis M. Lépilepsie myoclonique sévère de lenfant. Dans Journées Parisiennes de Pédiatrie. Paris: Flammarion, 1982:259-68.
- Nieto M, Roldán S, Sánchez B, Candau R, Rodríguez R. Estudio inmunológico en pacientes con epilepsia mioclónica severa en la infancia. Rev Neurol 2000;30:412-4.
- Nieto M, Candau R, Rufo M, Ruiz del Portal L. Intravenous immunoglobulin in severe myoclonic epilepsy of infancy. (Abstracts). Epilepsia 1996;37:102.
- Nieto M. Epilepsia mioclónica severa de la infancia (epilepsia polimorfa de la infancia). Rev Neurol 1994;22:143-6.
- Nieto-Barrera M, Candau R, Nieto-Jiménez M. Topiramato en el tratamiento de la epilepsia mioclónica severa de la infancia. Rev Neurol 2000;30:382-3.
- Chiron C, Marchand MC, Tran A, Rey E, dAthis P, Vincent J, et al. Stiripentol in severe myoclonic epilepsy in infancy: A randomised placebo-controlled syndrome dedicated trial. Lancet 2000;356:1638-43.
- Dalla Bernardina B, Fontana E, Sgró V, Colamaria V, Elia M. Epilepsie myoclonique (état de mal myoclonique) dans les encéphalopathies non progressives. En: Roger J, Bureau M, Dravet C, Dreifuss FE, Perret A, Wolf P, eds. Les syndromes épileptiques de l´enfant et de l´adolescent. 2e éd. London: John Libbey, 1992:89-96.
- Salas-Puig J, Calleja S. Epilepsias mioclónicas en el joven y en el adulto. Rev Neurol 1999;28:284-7.
- Delgado-Escueta AV, Greenberg DA, Teiman L, Liu A, Sparkes RS, Barbetti A, et al. Mapping the gene for juvenile myoclonic epilepsy. Epilepsia 1989;30:S8-18.
- Elmslie FV, Rees M, Williamson M, Kerr M, Kjeidsen MJ, Pang KA, et al. Genetic mapping of a major susceptibility locus for juvenile myoclonic epilepsy on chromosome 15q. Hum Mol Genet 1997;6:1329-34.
- Pedersen SB, Petersen KA. Juvenile myoclonic epilepsy: clinical and EEG features. Acta Neurol Scand 1998;97:160-3.
- Genton P, Salas-Puig X, Tuñón A, et al. Juvenile myoclonic epilepsy and related disorders: clinical and neurophysiological aspects. En: Malafosse A, Genton P, Hirsch E, Marescaux C, Broglin D, Bernasconi R, eds. Idiopathic generalized epilepsies. London: John Libbey, 1994:253-65.
- Appleton R, Beirne M, Acomb B. Photosensitivity in juvenile myoclonic epilepsy. Seizure 2000;9:108-11.
- Savic I, Lekvall A, Greitz D, Helms G. MR spectroscopy shows reduced frontal lobe concentrations of N-acetyl aspartato in patients with juvenile myoclonic epilepsy. Epilepsia 2000;41:290-6.
- Pellock JM. Treatment of seizures and epilepsy in children and adolescents. Neurology 1998;51:S008-S014.
- Mc Abee GN, Wark JE. A practical approach to uncomplicated seizures in children. Am Fam Physician 2000;62:1109-16.
- Gunatilake SB, Seneviratre SL. Juvenile myoclonic epilepsy: a study in Sri Lanka. Seizure 2000;9:221-3.
- Roger J, Genton P, Bureau M, Dravet C. Les épilepsies myocloniques progressives de l´enfant et de l´adolescent. En: Roger J, Bureau M, Dravet C, Dreifuss FE, Perret A, Wolf P, eds. Les syndromes épileptiques de l´enfant et de l´adolescent. 2e éd. London: John Libbey, 1992:381-400.
- Genton P. Últimos avances nosológicos y tratamiento de las epilepsias mioclónicas progresivas. Rev Neurol 1999;28:269.
- Vistorte A, Sardiñas N, Esteban EM, Vargas-Díaz J, Novoa-López L, Rojas E, et al. Epilepsia mioclónica progresiva: caracterización clínica de 18 pacientes. Rev Neurol 1999;29:102-4.
- Mrabet A, Gargouri A, Fredj M, Gouider R. EEG in progressive myoclonic epilepsy type Unverricht-Lundborg Disease. (Abstract). Epilepsia 2001;42(suppl 2):104.
Recibido: 30 de enero de 2001. Aprobado: 6 de marzo de 2001.
Dra. Albia J. Pozo Alonso. Servicio de Neuropediatría. Hospital Pediátrico Universitario «William Soler», San Francisco y Perla, 10800, Altahabana, municipio Boyeros, Ciudad de La Habana, Cuba. Correo electrónico. albiap@infomed.sld.cu
1 Neuropediatra.
2 Doctor en Ciencias Médicas. Neuropediatra. Profesor Titular de Pediatría.
3 Residente de Medicina General Integral. Policlínico Capdevila, municipio Boyeros. Ciudad de La Habana.

