










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Pediatría
versión On-line ISSN 1561-3119
Rev Cubana Pediatr v.73 n.4 Ciudad de la Habana oct.-dic. 2001
Hospital General Provincial Docente Carlos Manuel de Céspedes, Bayamo, Granma
Síndrome de Freeman-Sheldon. Revisión bibliográfica
Dr. Manuel Estrada Sarmiento,1 Dra. Alicia La O Cabrera,2 Lic. Isel Virelles Espinosa,3 Dra. Sara Ferrándiz Guerra4 y Dr. Elpidio Ortiz Castellanos4
Resumen
Se realiza una revisión bibliográfica actualizada sobre las peculiaridades clínicas de la displasia craneocarpotarsal o síndrome de Freeman-Sheldon. Los hallazgos característicos de este síndrome se presentan en el nacimiento, con predominio de anomalías musculoesqueléticas, cuyos signos relevantes fueron la microstomía, la hipoplasia del tercio medio de la cara y el surco en forma de H en el mentón, signo patognómico. El diagnóstico de esta afección se efectúa por los hallazgos faciales y de los miembros; de mucha utilidad resultaron los estudios radiográficos de cráneo, la biopsia del músculo buccinador y los estudios electromiográficos. El tratamiento quirúrgico se indica para mejorar la apariencia facial y la función de las manos y de los pies. Se profundiza en los aspectos clínicos, diagnóstico y terapéuticos de esta enfermedad.
DeCS: HUESOS FACIALES/anomalías; HUESOS DE LA EXTREMIDAD SUPERIOR/anomalías; DEFORMIDADES CONGENITAS DEL PIE/fisiopatología.
El síndrome de Freeman-Seldon, conocido también como síndrome de la cara silbante y displasia craneocarpotarsal,1-6 se describió por ambos autores en 1938. Ciertas malformaciones esqueletales con características variables se asocian con este síndrome. Hasta el momento de nuestra revisión, solo se habían reportado en el mundo 50 casos. Etiología El defecto básico se desconoce, aunque la mayoría de los casos es de presentación esporádica, existen bastantes indicios de que se trata de una hernia autosómica dominante.1,7-11
Características clínicas
Este síndrome se caracteriza por alteraciones en cráneo, cara, extremidades y otros síntomas. Los hallazgos característicos de este trastorno se presentan al nacimiento.11-20
Cara y cráneo (figs. 1 a y 1 b)
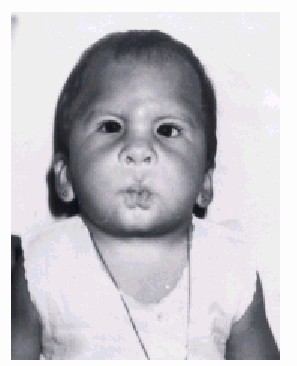
Fig. 1a. Vista de frente donde se aprecian las características en cara y cráneo del síndrome.
- Frente alta.
- Facies semejante a una máscara.
- Hipoplasia del tercio medio de la cara.
- Hipotelorismo.
- Pliegues epicántricos.
- Ptosis del párpado superior.
- Inclinación antimongoloide de las hendiduras palpebrales.
- Estrabismo convergente.
- Blefarofimosis.
- Philtrum largo.
- Boca pequeña que da una apariencia de silbador.
- Pliegue en forma de H en la barbilla.
- Paladar ojival.
- Maxilar inferior y lengua pequeña.
Extremidades superiores (fig. 2)
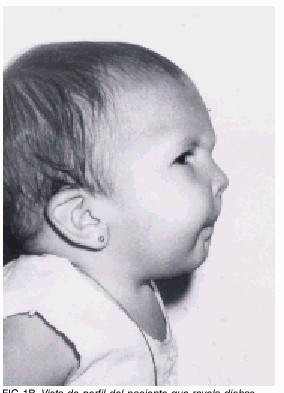
1b. Vista de perfil del paciente que revela dichas características sindrómicas.
- Miembro superior con escasez de masa muscular.
- Limitación de la movilidad de los hombros.
- Disminución de la pronación y supinación de los antebrazos.
- Deformidades de los pulgares en el nivel de la articulación metacarpofalángica.
- Desviación cubital de los dedos sin que existan anomalías óseas.
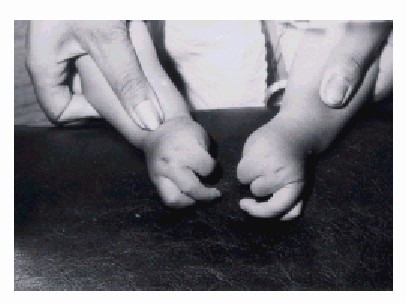
Fig. 2. En la figura se destacan las malformaciones de los miembros superiores.
Extremidades inferiores (fig.3)
- Pie equinovaro bilateral.
- Pie en mecedora.
- Tobillo vertical.
Otras
- Deficiente crecimiento posnatal.
- Cuello corto con pteriglon.
- Escoliosis, cifosis.
- Espina bífida oculta.
- Dislocación de la cadera.
- Hernia inguinal.
- Dificultad para la deglución.
- Vómitos.
- Laringomalacia.
Diagnóstico
Los hallazgos faciales y de los miembros en este síndrome permiten fácilmente el diagnóstico, el cual se realiza por los siguientes aspectos:4,5,21-25
Radiografía de cráneo
Se observa apariencia anormal del suelo de la fosa craneal media.
Biopsia del músculo buccinador
Los haces musculares se encuentran reemplazados por tejido conectivo fibroso.
Estudios electromiográficos
Se aprecia disminución de la actividad más acentuada en los músculos de la expresión facial.
Diagnóstico diferencial Se realizará el diagnóstico diferencial con los siguientes síndromes:5,26,27
- Síndrome de Schwart-Jample: Cara inmóvil por contracción tónica de los músculos faciales, labios fruncidos, pectus carinatun, displasia de cadera y contractura articulares.
- Trastornos autosómicos dominantes: Se caracterizan por incapacidad para abrir la boca, seudocamptodactilia y cortedad de tibia.
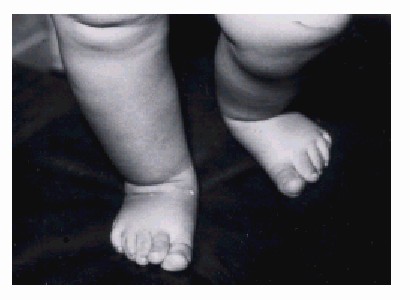
Fig. 3. Se revela la apariencia anormal de los miembros inferiores.
Tratamiento
Se indica la cirugía plástica y ortopédica para mejorar la apariencia facial y función de la mano y de los pies.5,16,27-30
Microstomía
La corrección se deberá efectuar en un tiempo. Reconstruir una comisura correctamente exige mucho cuidado. La intervención se debe realizar después del año; el paciente reúne mejores condiciones para el acto quirúrgico y los tejidos perilabiales han aumentado de tamaño, lo que facilitará así una cirugía de precisión.29
Malformaciones de miembros superiores e inferiores
Para la desviación cubital se utilizan férulas para corregir la deformidad: si no resuelve se emplea el tratamiento quirúrgico. En el pie equinovaro de no ser satisfactorio el tratamiento conservador, el quirúrgico se realizará entre los 6 meses y 1 año de edad.
Pronóstico
En esta afección tanto la inteligencia como el promedio de vida son normales en la época de lactancia. Los vómitos y los trastornos de la deglución pueden conducir a un retraso pondoestatural.5,27,31
Summary
An updated bibliographic review on the clinical pecularities of craniocarpotarsal dysplasia or Freeman-Sheldon syndrome was made. The characteristic findings of this syndrome were observed at birth, with predominance of musculoskeletal anomalies, whose significant signs were microstomia, hypoplasia of the median third of the face and the H-shaped sulcus in the chin, a pathognomic sign. The diagnosis of this affection is made by using the facial and extremity findings. The radiographic studies of the cranium, the biopsy of the buccinator muscle and the electormiographic cranial studies proved to be very useful. The surgical treatment was indicated to improve the facial look and the function of the hands and feet. The clinical, diagnostic and therapeutic aspects were analyzed in detail.
Subject headings: FACIAL BONES/anomalies; BONES OF UPPER EXTREMITY/anomalies; FOOT DEFORMITIES, CONGENITAL/physiopathology.
Referencias bibliográficas
- Patel M, Mahonud F. A case of Freeman-Sheldon syndrome (Cranio-carpotarsal-dysplasia). Br J Radiol 1999;76(663):80-83.
- Hirschfelder U. Craniocarpotarsal dystrophy (Freeman-Sheldon syndrome). Dtsch Sahnarztl 1998;45(4):209-12.
- Guzzanti Y, Toniolo RM. The Freeman-Sheldon syndrome Arch Putti Chir Organi 1998;38(1): 215-22.
- Parisi G Molino O Freeman-Sheldon syndrome. Case contribution and review of the literature. Minerva Pediatr 1999;43(10):653-9.
- Goodman RM. Malformaciones en el lactante y en el niño. Madrid: Salvat; 1986. p. 244-5.
- Susanes Cabello A, Espino Aguilar B. Freeman Sheldon syndrome. Report of a case. An Esp Pediatr 2000;36(4):256-9.
- Wang TR. Further evidence for genetic heterogeneity of whisthing face or Freeman –Sheldon syndrome in a Chinese family. Am J Med Genet 1998;38(2):456-9.
- Bonioli E, Bellini C, Rufa G. Freeman-Sheldon syndrome. Description of 2 cases of probable recessive autosomal inheritance. Minerva Pediatr 1999;38(3-4):143-9.
- Sánchez JM. New evidence for genetics heterogeinity of the Freeman-Sheldon syndrome. Am J Med Genet 1999;25(3):508-12.
- Vanck J; Janda J, Losan P. Freeman-Sheldon: A disorder of congenital myopathia origin. Can Anesth Soc J 1998;33(3):388-93.
- Alves AF. Reccesive foron of Freeman-Sheldon syndrome or Whistling Face. J Med Genet 1999;14(1):39-41.
- Aldinger G, Euler TJ. The Freeman-Sheldon syndrome. Z Orthop Hre Grenzgeb 1999;121(5):730-9.
- Goyel NA, Patel Zon, Wagle PR. Freeman-Sheldon syndrome. Indian Pediatr 2000;32(4):153-9.
- Cirillo Silengo M, Davi GF, Bianco R. The Freeman- Sheldon syndrome with mental retardation. Minerva Pediatr 1999;36(6):280-9.
- Estrada R. Freeman -Sheldon syndrome with unusual hand and foot anomalies. J Nati Med Assoc 2000;81(6):764-8.
- Krakowiak PA, Carey JC. Clinical analysis of a variant of Freeman-Sheldon syndrome. Am J Med Genet 2000;86(5):90-9.
- Bekir N, Coskum Y. Whistling face syndrome in two siblings. Turk J Pediatr 1998;36(4):329-32.
- Zampino G, Conti G, Balducci F. Severe form of Freeman-Sheldon syndrome associated with brain anomalies. Am J Med Gen 1999;62(3):290-7.
- Song HB, Sarwark Grant J. Freeman -Sheldon syndrome and cranio vertebral junction malformation producing dysphagia and weight loss. Pediatr Neurosurg 2000;20(2):172-6.
- Phadke S, Sharma A, Freeman-Sheldon syndrome with bilateral simian crease and malpositioned second toes. Indiam Pediatr 1999;30(1):90-6.
- Antlex PM. Diagnostic criteria for the whistling face syndrome. Cith Defects 1996;11(5):160-8.
- Robins Furman F. Prenatal diagnosis of Freeman-Sheldon syndrome. Prenat Diagn 1998;25(1-3): 243-8.
- Sobrado CG, Ribera M, Marti M. Freeman-Sheldon syndrome: generalized rigidity. Rev Esp Anesthesiol Reanim 1998;41(3):180-4.
- Galliani CA. Laryngomalacia and intraneural striated muscle in an infant with the Freeman-Sheldon syndrome. Int J Pediatr Otorhinolaryngol 1993;25(1-3):243-8.
- Manji KP, Mbise RL. Generalized muscle hypertomia with mask – life face in a Tanzanian giol. Clin Genet 1998;54(3):252-3.
- Simosa V, Bustos T. A new syndrome with distinct facial and auricular malformations and dominant inheritance. Am J Med Genet 1999;32(2):184-6.
- Mckusick VA. Mendelian inheritance in man. Baltimore: The John Hopkins University Press; 1988.
- Ohyama K; Susami t, Kato Y. Freeman-Sheldon syndrome: case management from age la to 16 years. Cleft Palate Craniofac 1999;34(2):151-3.
- Ferreira LM, Mirami E, Andrews JD. Freeman-Sheldon syndrome: surgical correction of microstomia. Br J Plast Surg 1998;47(3):201-2.
- Martini AK. Surgical treatment of the hand deformity in Freeman-Sheldon syndrome. Randchir Mikrochir Plast Chir 1997;14(6):210-9.
- Nara T. Reconstruction of an upper lip and the coloboma in the nasal ala, accompanying with Freeman-Sheldon syndrome. Nippon Geka Hokan 1987;50(11):626-32.
Recibido: 26 de diciembre del 2000. Aprobado: 7 de marzo del 2001.
Dr. Manuel Estrada Sarmiento. Máximo Gómez, número 6, entre Maceo y Canducha Figueredo, Bayamo, CP 85100, Granma, Cuba.
1 Especialista de II Grado en Cirugía Maxilofacial. Profesor Auxiliar. Jefe del Grupo Provincial de Maxilofacial. 2 Especialista de I Grado en Genética Clínica. 3 Licenciada en Enfermería. Jefe de Enfermeras de la Salud de Cirugía. 4 Especialista de I Grado en Pediatría. Asistente.

