










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Pediatría
versión impresa ISSN 0034-7531versión On-line ISSN 1561-3119
Rev Cubana Pediatr v.77 n.3 Ciudad de la Habana jul.-dic. 2005
Hospital Docente Ginecoobstétrico «América Arias»
Síndrome de Beckwith Wiedemann. Presentación de un caso
Dr. Eugenio Carro Puig1 y Dra. Liliam S. Fernández Braojos2
RESUMEN
Se presenta el caso de una paciente atendida en el Servicio de Neonatología del Hospital Pediátrico «Arthur Davison» de Ndola (Zambia). La paciente es portadora de un síndrome de Beckwith Wiedemann, enfermedad de posible origen genético autosómico dominante, cuyo diagnóstico se fundamenta en criterios clínicos y de laboratorio. Se exponen los resultados del examen clínico y de las investigaciones complementarias. Se realiza una revisión de la literatura médica sobre el tema.
Palabras clave: Convulsiones refractarias, onfalocele, macroglosia, síndrome de Beckwith Wiedemann.
El primer reporte de una asociación causal entre la macroglosia, el onfalocele y otras anomalías umbilicales y la hipoglicemia sintomática fue hecho en 1963 por Beckwith.1-3 Posteriormente esta asociación ha sido descrita por otros autores en la literatura médica.4-6
Se estima que el síndrome puede presentarse aproximadamente en 1 de cada 13 700 nacimientos, aunque es posible que no sean diagnosticados los casos menos severamente afectados. La incidencia en uno y otro sexo es similar y la frecuencia de presentación igual en individuos de la raza blanca o negra.
En algunos estudios citogenéticos se reportan alteraciones en el cromosoma 11 (11p15.5). El antecedente de consanguinidad en los padres sugiere un patrón de herencia autosómica recesiva. En la actualidad, se describe como causa de este síndrome la disomía uniparental.
Por lo general se ha reportado en neonatos cuyo peso los clasifica como grandes según la edad gestacional. La presencia de hipoglicemia sintomática intensa persistente está asociada a hiperplasia difusa de los islotes de Langerhans e hiperinsulinemia.3-6 Junto a la macroglosia y onfalocele típicos en la enfermedad, se pueden encontrar visceromegalia (hiperplasia de hígado y riñones) y hemihipertrofias.
Estos enfermos poseen una tendencia aumentada respecto a la población genera, de desarrollar neoplasias, como carcinomas renales, tumores de Wilms y nefroblastomas bilaterales.7-8
La existencia de las anomalías descritas y el trastorno metabólico son característicos de la enfermedad, por lo que la presencia de estos elementos permite hacer el diagnóstico positivo de esta. En casos similares debe hacerse el diagnóstico diferencial con respecto al síndrome de Simpsom-Golabi-Behmel, al síndrome Perlman y al síndrome Costello. Los exámenes genéticos tienen valor para confirmar el diagnóstico, definir el riesgo y hacer diagnósticos prenatales.
PRESENTACION DEL CASO
En el Servicio de Neonatología del Hospital Pediátrico «Arthur Davison» se recibe una recién nacida del sexo femenino, raza negra, con peso de 4 200 g, procedente del Hospital Central de Ndola, por presentar onfalocele y convulsiones refractarias al tratamiento convencional.
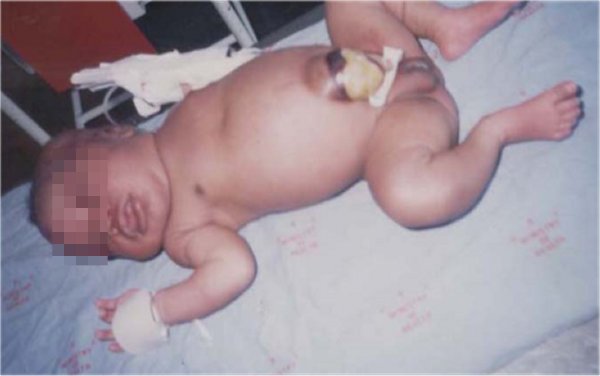
Figura. Nótese los signos físicos: onfalocele, macroglosia, etc.
En los antecedentes se recoge nacimiento por parto eutócico, con Apgar 9-9, de una madre con dos partos anteriores normales. No se pudo conocer de antecedentes familiares de trastornos similares o sobre la existencia de consanguinidad entre los padres.
Momentos después del nacimiento, la paciente comenzó a convulsionar, por lo que recibió tratamiento con anticonvulsivantes del tipo del fenobarbital, el diazepam y el convulsín, con los que no se logró solucionar los síntomas y se decide el traslado hacia nuestra institución, pues tiene una capacidad resolutiva superior.
Se realiza una exploración física exhaustiva y se comprueba la presencia de macrosomía, convulsiones, onfalocele, macroglosia y hepatomegalia. Se le realizan investigaciones de laboratorio, tales como hemograma, estudio de líquido cefalorraquídeo (LCR) y glicemia, y se detecta una glicemia de 1,8 mol/L. El resto de los complementarios mostró resultados normales.
Se continuó el tratamiento anticonvulsivo con fenobarbital y se intentó la corrección de la hipoglicemia por bolo. Para lograr la estabilización de cifras adecuadas de glicemia fue necesario mantener infusión endovenosa de dextrosa cuyo flujo se llevó a 10 mg (kg/min), así como adicionar hidrocortisona a la terapéutica. Esta última permitió reducir las concentraciones de dextrosa y finalmente retirar la infusión. El esteroide se mantuvo hasta el séptimo día de vida. La paciente se restableció de las convulsiones cuando se normalizaron las cifras de glicemia en sangre.
Se remitió entonces a otro centro para su seguimiento por Endocrinología y posible corrección del onfalocele.
DISCUSIÓN
El diagnóstico del síndrome de Beckwith Wiedemann se basa clásicamente en el hallazgo de macroglosia, onfalocele u otras anomalías umbilicales acompañadas de hipoglicemia sintomática persistente, que se presenta en los neonatos macrosómicos habitualmente sin otros antecedentes perinatales de interés.
El desarrollo de la hipoglucemia sintomática aparece poco después del nacimiento y las convulsiones provocadas por ésta se hacen refractarias al tratamiento anticonvulsivo si no se corrigen las cifras de glicemia en sangre, como cabe esperar.
Nuestra paciente reúne las características descritas en la literatura médica, ya que era portadora de las anomalías que se señalan y del trastorno metabólico planteado, el cual le ocasionó un cuadro convulsivo refractario al tratamiento convencional. Como dato adicional, existían antecedentes familiares de consanguinidad.
Las alteraciones anatómicas observadas, así como del trastorno metabólico, los antecedentes de consanguinidad, la forma de inicio del cuadro y la respuesta al tratamiento aplicado hacen posible el diagnóstico diferencial con respecto a otros trastornos convulsivos y afecciones en las que se presentan anomalías congénitas como las encontradas en la paciente.
Por todas las razones expuestas, se concluyó que la paciente presenta un síndrome de Beckwith Wiedemann.
AGRADECIMIENTOS
Queremos agradecer a la Revolución, que nos permitió brindar nuestros servicios médicos en la República de Zambia, donde tuvimos la oportunidad de salvar muchas vidas y la experiencia de observar enfermedades poco frecuentes, como la que se presenta.
REFERENCIAS BIBLIOGRÁFICAS
1. Roberton NRC. Texto de Neonatología. Massachussets: Universidad de Cambrigde; 1986. pp. 612, 613.
2. Fanaroff-Martin. Enfermedades del feto y el recién nacido. 5ta Edición. La Habana: Ed. Científico-Técnica; 1985. pp.1038-40.
3. Sola A, Rogido M. Cuidados Especiales del Feto y del Recién Nacido. 2a ed, Vol 1. Buenos Aires: Marcelo T. De Alvear Interamericana; 2001. p.192.
4. Beckwith Wiedemann Reviewed by Adam Editorial. 2002. Available from: www.beckwith-wiedemann.org/
5. Shuman Ch, Weksberg R. Beckwith Wiedemann Syndrome. Gene Reviews. 2003. Available from: www.gentests.org
6. Nelson. Tratado de Pediatría.15 ed. Madrid: Mc Graw-Hill, Interamericana; 1997. pp. 532 y 642 .
7. Kissane JM, Denhner LP. Renal tumors and tumors-like lesions in pediatric patients. Pediatr Nefrol. 1992; 6:365-82.
8. Suchida Y, Yokomori K, Choi SH. Hereditary renal tumors: Wilms´tumor congenital anomalies syndrome. Nippon Rinsho. 1995; 53(11):2742-8.
Recibido: 4 de marzo de 2004. Aprobado: 15 de agosto de 2005.
Dr. Eugenio Carro Puig. Calle Línea y Avenida de los Presidentes, Vedado. Ciudad de La Habana, Cuba.
Correo electrónico: ecarro@infomed.sld.cu
1Especialista de I Grado en Neonatología.
2Especialista de I Grado en Neonatología y Medicina General Integral.

