










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Pediatría
versión impresa ISSN 0034-7531versión On-line ISSN 1561-3119
Rev Cubana Pediatr v.78 n.2 Ciudad de la Habana abr.-jun. 2006
Presentación de casos
Hospital General Docente «Iván Portuondo», San Antonio de los Baños (La Habana)
Higroma quístico del cuello
Dra. Yanet García Fernández,1 Dra. Rosa Maria Fernández Ragi2 y Dr. Jorge Luis Russinyol Nieto3
RESUMEN
Se presenta el caso de un recién nacido, nacido fallecido, con diagnóstico prenatal de higroma quístico obtenido mediante ultrasonografía intrauterina. Se describen los hallazgos clínicos, se revisa la literatura sobre el tema, se hace énfasis en los diagnósticos diferenciales clínicos y anatomopatológicos de esta enfermedad infrecuente. Se valora la importancia del diagnóstico prenatal y del Programa Nacional de Genética en nuestro país.
Palabras clave: Linfangioma, higroma quístico.
Con la instalación y aplicación de los programas de genética en nuestro país cada día resulta menos posible al nacimiento la aparición de malformaciones en el neonato, pues estas son diagnosticadas intraútero. No obstante ello, algunas escapan a los controles prenatales. No es lo que ocurrió en el caso que hoy exponemos, pues este fue diagnostico por ultrasonografía en el ultimo trimestre del embarazo.
El higroma quístico es una malformación congénita, que consiste en uno o más espacios linfáticos llenos de líquido. Se puede observar en recién nacidos sanos y está asociado a trastornos de tipo genético.1 Su incidencia es relativamente baja, aproximadamente de 1 por 50,000 nacimientos.2
Por la baja incidencia de esta enfermedad y por la posible contribución a la literatura médica, decidimos presentar nuestro caso.
PRESENTACION DEL CASO
Se trata de un recién nacido con antecedentes de madre de 30 años de edad, segundo embarazo procedente de medio rural, que durante el embarazo presentó infección urinaria. No existen otros antecedentes familiares ni del embarazo.
En el último trimestre del embarazo se diagnostica por ultrasonografía una tumoración en el cuello del feto y se pone a la madre en trabajo de parto espontáneo 1 semana después del ultrasonido. Transcurría la semana 32 de la gestación, no se había concluido el diagnóstico prenatal y la embarazada parió un feto pretérmino de bajo peso, fallecido. Al examen físico se constata una tumoración lobulada en el cuello, grande, con localización en zonas laterales del cuello y extensión al centro, de consistencia blanda. El peso del feto fue de 2 000 g. En el estudio anatomopatológico se comprueba que dicha tumoración se corresponde a un higroma quístico del cuello.
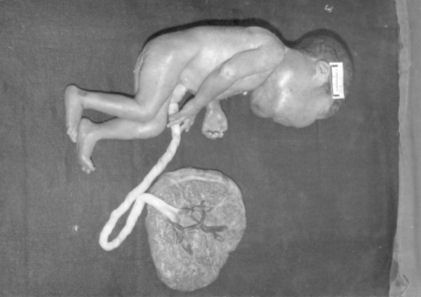
Figura 1. Se obsesa en la foto la gran tumoración bilateral en el cuello del feto.
DISCUSIÓN
Aparecen en la literatura algunos reportes de casos diagnosticados con esta enfermedad. Se plantea una incidencia variable por algunos autores,2 pero la incidencia planteada es de 1 por cada 50,000 nacidos vivos.3,4 Nuestra incidencia fue de l por 60,000 nacidos vivos, aunque nuestro caso falleció intraútero. Encontramos una tumoración grande bilateral, cuya consistencia fue blanda y que presentaba compromiso compresivo de vías respiratorias y vasos del cuello. No se observaron otras malformaciones aparentes clínicamente ni desde el punto de vista anatomopatológico.
Resulta interesante conocer acerca de esta entidad clínica planteada con una frecuencia variable. En el Hospital Sarda (Argentina) se han diagnosticados en forma continuada cinco casos,2 de los cuales uno solo se diagnosticó intraútero y nació muerto. Así observamos casos que son diagnosticados después del nacimiento como una masa tumoral mal definida, de poca consistencia, multilobulada y fluctuante. Está formada por múltiples quistes de diferentes tamaños, constituidos a expensas de los vasos linfáticos del cuello que se desordenan, y en ocasiones con un componente vascular venoso asociado (hemangiolinfangiomas).2 El crecimiento es progresivo y lento en los primeros meses de vida, contienen un líquido claro y transparente y puede presentar exacerbaciones secundarias a infecciones, hemorragia o sin explicación aparente, y también producir trastornos respiratorios y circulatorios por compresión.
Sin distinción entre sexos, el higroma quístico o linfangioma es una malformación inespecífica de los canales linfáticos. Se plantea que es una anomalía congénita de los vasos linfáticos yugulares, que trae como consecuencia el drenaje no adecuado de la linfa.
El linfangioma se desarrolla a partir de los sacos linfáticos secundarios cuando son secuestrados por los primarios durante la vida embrionaria. Se propone que esta malformación es el resultado de segmentos del saco linfático yugular que están fuera de sitio o de la falla de los espacios linfáticos para conectar con los principales canales linfáticos y constituye un tumor líquido claro, limpio y transparente.1,2
En el momento del nacimiento o posteriormente, el diagnostico es clínico. El higroma quístico puede diagnosticarse con seguridad por ecografía en el segundo trimestre del embarazo. Se identifica como una masa que surge del cuello posterior o lateral y puede diferenciarse de otras causas (encefalocele posterior o meningocele occipital, mielomeningocele, teratoma quístico benigno, quiste subcoriónico placentario, edema de la nuca, hemangioma, teratoma o cordón umbilical) por un cráneo y columna íntegra, falta de componente sólido, posición constante respecto a la cabeza fetal y presencia de cavidad y septo dentro de la masa. El higroma quístico también se asocia con linfedema (69 %), hidrops (46 %), oligoamnios (68 %) y otras anomalías, como retardo de crecimiento intrauterino, cardíacas, polihidramnios, etc. En todos los casos existe disminución o ausencia de movimientos fetales. No hay tratamiento fetal para el higroma quístico y el mejor método de investigación citogénica es la biopsia del vello coriónico, y está indicado, en los lugares si es permitido, la interrupción del embarazo.
En el mayor porcentaje de los casos se ubica en la región lateral del cuello, detrás del músculo esternocleidomastoideo y pueden penetrar el músculo. Se extienden al tejido celular subcutáneo del triángulo posterior de la nuca y en ocasiones puede ubicarse en el área subglótica, submaxilar, nuca, axila, mediastino, retroperitoneal e inguinal. Los quistes pueden exceder los 5 cm. de diámetro y pueden estar comunicados unos con otros o permanecer aislados. Tienen a a ser bilaterales, situados lateralmente a las venas yugulares internas. Dichas ubicaciones guardan relación con la embriogénesis, ya que en el final de la novena semana de la gestación comienza a desarrollarse el sistema linfático, análogamente al sistema venoso, para luego separarse de este y formar 5 sacos linfáticos: uno retroperitoneal simple y 2 pares cerca de la vena yugular y ciática respectivamente.
Se sugiere que está frecuentemente asociado a anomalías cromosómicas y que es más frecuente en el síndrome de Turner y menos frecuente en la trisomias 13,18,21, síndrome de Klinelfelter, síndrome de Noonan, Cowchock, Cunning y Roberts. Además pueden verse en otro tipo de malformaciones no genéticas como los síndromes de alcoholismo fetal, aminopterina fetal y trimetadiona fetal.2
Desde el punto de vista clínico estos tumores se presentan como una masa cervical quística única o múltiple, de tamaño variable, consistencia blanda, indolora, mal delimitada, fluctuante, lobulada, multitabicada, translúcida, no adherida a tejidos profundos y la piel que lo cubre puede ser delgada y de color azul. Su crecimiento es lento y progresivo a lo largo del primer año de vida. Cuando no hay infección, las paredes de los quistes son delgadas, de color perlado, casi transparentes y contienen un líquido claro. En ocasiones se produce la ruptura de algunos quistes, con hemorragia asociada y se confunde con un linfohemangioma.3-5
En general los síntomas están relacionados con la localización del quiste y su tamaño. Si la masa quística comprime la vía aérea, ocasiona síndrome de dificultad respiratoria y trastornos de la deglución. Cuando está localizado en la región perifaríngea puede ocasionar alteraciones en la función de la articulación temporo-mandibular. La masa supraclavicular se puede evidenciar al realizar la maniobra de Valsalva y en estos casos el higroma está asociado a una localización mediastínica.5-7
El diagnóstico es fundamentalmente clínico y la transiluminación orienta hacia el contenido líquido del tumor. La ecografía cervical, radiografía de tórax y tomografía axial computarizada ayudan a evaluar la extensión y características. Prenatalmente se puede encontrar una disminución de los niveles de alfafetoproteína y aumento de la fosfatasa alcalina en suero materno.
En el período neonatal, el diagnóstico diferencial debe hacerse con otras tumoraciones como son: teratoma quístico benigno, hemangiomas, anomalías del arco braquial y del conducto tirogloso, tortícolis congénita; y en el niño mayor con linfoadenitis, neurofibromas, tumores salivales, tumores de tiroides, linfoma de Hodking, neuroblastoma cervical, rabdomiosarcoma, leucemia, histiocitosis X.
Las complicaciones más frecuentes son la hemorragia e infección sobreagregada. Es de buen pronóstico el hecho de poder realizar la resección total de la masa tumoral, si otros órganos no están comprometidos o si no se asocian otras malformaciones como señalamos anteriormente.
En el tratamiento la resección total del tumor es lo fundamental, pero en la mayor parte de los casos esto es imposible, porque infiltra estructuras como la lengua y faringe o está adherido a estructuras vitales como el plexo cervical, nervio frénico, nervio vago, vena yugular, arteria carótida, en este caso es necesario la utilización de la radioterapia, corticoides, bleomicina como agentes esclerosantes, aunque la mayoría de las veces estos no son efectivos. En el excepcional caso de que el higroma esté constituido por un quiste simple, o a lo sumo dos, se puede antes de indicar el tratamiento quirúrgico intentar la absorción del quiste y su sellado posterior con un pegamento biológico denominado Tisusucol.8
El tratamiento quirúrgico dependerá de si el paciente presenta complicaciones o no y debe de realizarse antes del año de edad. Esta patología puede ser un marcador de mal pronóstico cuando se diagnostica en el segundo trimestre de embarazo. El higroma simple en el primer trimestre aparenta tener mejor pronóstico; los fetos con higroma del primer trimestre tienen alto riesgo de aneuplodía cromosómica y se les debería realizar estudio prenatal citogenético. En aquellos que presentan cariotipo normal y no están asociados a hidrops, es esperable resolver el higroma sin que queden secuelas.
Cuando un higroma quístico es detectado por ecografía fetal, está indicado un estudio ecográfico cuidadoso para anomalías asociadas; así, deberían pensarse en características sugestivas de síndrome de Turner (síndrome de corazón izquierdo hipoplásico, coartación de aorta, riñón en herradura), síndrome de Down (atresia duodenal, canal AV), síndrome de Roberts. La presencia de higroma quístico, sin complicaciones severas, implica una supervisión ecográfica cuidadosa y de pronóstico reservado. Una supervisión multidisciplinaria integrada por obstetras, radiólogos, neonatólogos, genetistas, cirujanos y asistentes sociales, está indicada cuando los antecedentes son inciertos. El tratamiento en el período postnatal es quirúrgico, y está indicado entre los 4 a 12 meses de vida. La compresión de las vías aéreas o las infecciones recurrentes pueden obligar a realizar la intervención en una edad más temprana.
No es necesario sacrificar nervios o estructuras vasculares para lograr la escisión total de esta lesión benigna y son preferibles las intervenciones repetidas múltiples del higroma residual. La principal complicación después del tratamiento quirúrgico es la recidiva de los quistes, ya que pueden existir microquistes difícilmente identificables. En los últimos años se está adquiriendo experiencia con el uso de sustancias esclerosantes de los vasos linfáticos que forman el higroma, esto es, sustancias que se inyectan localmente y logran el cierre de los quistes y la desaparición de estos. Parece ser más eficaz cuando los quistes son grandes que cuando se trata de microquistes, y puede convertirse en la primera elección terapéutica en los niños con estas malformaciones linfáticas. La más usada de estas sustancias es el OK 470, pero hoy en día es una sustancia cara y no disponible en las farmacias, y solo puede utilizarse siguiendo un protocolo hospitalario estricto.9 Otra de las sustancias utilizadas es el OK 432, que es una mezcla incubada liofilizada de Streptococcus pyogenes del grupo A, de origen humano, que produce un aumento en el número de neutrófilos, macrófagos y leucocitos en el fluido del higroma quístico, además aumenta el número de células asesinas y linfocitos T, con mayor concentración de factor de necrosis tumoral y de interleucina.
Todos estos fenómenos aumentarían la permeabilidad del endotelio, y así el drenaje acelerado del contenido, que produciría la contracción de los espacios quísticos.9,10 Finalmente, la atención de las madres de estos niños debe estar encaminado en el futro hacia un consejo genético adecuado.
REFERENCIAS BIBLIOGRÁFICAS
1. Langman J. Embriología médica. 7a ED. México: Editorial Médica Panamericana; 1996. pp. 292-324.
2. Brunori EA, Caratozzolo G, Martínez M, Dinerstein NA. Higroma Quístico. Presentación de un caso. Rev. Hosp. Mat. Inf. Ramón Sardá. 1996;15:(2)94-6.
3. Ogita S, Tsuto T, Nakamura K. OK-432 Therapy for lymphangioma in children: why and how does it work? J Pediatric Surg. 1996; 31: 477-480.
4. Barriga J, Murillo C, Agreda JA. Higroma Quístico, a propósito de un caso. Rev Boliviana de Pediatría. 2002; 41(2):1-4.
5. Edwards MJ, Graham JM. Posterior nuchal cystic hygroma. Clinics in Perinatology. 1990;17: 611-635.
6. Kalousek DK, Seller MJ. Differential diagnosis of posterior cervical hygroma in previable fetuses. Am J Med Genet Suppl. 1987; 3: 83-92.
7. Núñez Acevedo N. Salud infantil. Temas de Pediatría. 2003.[En línea] Consultado el 16 de noviembre de 2005. Disponible en: http://www.medynet.com
8. Feldman DS, Nevelon-Chevalier A, Degrolard M, Harran MH, Jahier J. Retrocervical Cystic Hygroma: diagnosis, prognosis and management a series of 13 cases. J. Gynecol Obst Biol Reprod. 1991; 20:183-90.
9. Pantoja M, Mazzi E. Higroma quístico cervical (linfangioma). Rev. Soc Bol Ped. 1999; 38(11):8-9.
Recibido: 27 de febrero de 2006. Aprobado: 16 de marzo de 2006.
Dra. Yanet García Fernández. Calle 78 N.o 3310, e/ 33 y 35, San Antonio de los Baños.
Correo electrónico: yanet.fernandez@infomed.sld.cu
1 Especialista de I Grado en Medicina General Integral y Neonatología. Profesor Instructor.
2 Especialista de II Grado en Neonatología. Profesor Auxiliar.
3 Especialista de I Grado en Ginecología y Obstetricia. Profesor Asistente.

