










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Pediatría
versión On-line ISSN 1561-3119
Rev Cubana Pediatr v.80 n.1 Ciudad de la Habana ene.-mar. 2008
PRESENTACIÓN DE CASOS
Duplicación 2 q 2.1- q 3.1: reporte de un caso
Duplication 2 q 2.1- q 3.1: a case report
Lic. Marta M. Acosta Sabatés,I Dr. Iván Hernández García,II Dra. Débora A. García Martínez,III y Dra. Kalia Lavaut SánchezIV
ILicenciada en Ciencias Biológicas. Máster en Antropología. Hospital Pediátrico Universitario «William Soler», Servicio de Genética. La Habana, Cuba.
IIEspecialista de I Grado en Genética Clínica. Hospital Pediátrico Universitario «William Soler». La Habana, Cuba.
IIIEspecialista de II Grado en Neonatología. Máster en Atención Integral al Niño. Hospital Pediátrico Universitario «William Soler». La Habana, Cuba.
IVEspecialista de I Grado en Genética Clínica. Instituto de Hematología e Inmunología. La Habana, Cuba.
RESUMEN
Se presentan las características clínicas y el estudio cromosómico de un paciente con trisomía parcial 2q 2.1-q 3.1. El lactante presentaba hendiduras palpebrales antimongoloides, labios finos, microtia bilateral, ojos profundos y microrretrognatia, entre otras dismorfias. El cuadro malformativo incluía cataratas, microftalmía, coartación de la aorta y asimetría de los ventrículos laterales del cerebro. Se le realizó estudio citogenético, cuya formula cromosómica resultante fue 46, XY, dup (2) (q 2.1-q 3.1). El paciente falleció a los 5 meses de edad como consecuencia de una neumonía complicada.
Palabras clave: Aberración cromosómica estructural, duplicación 2q 2.1- q 3.1, trisomía parcial 2q 2.1 - q 3.1.
SUMMARY
The clinical characteristics and the chromosomal study of a patient with partial trisomy 2q 2.1-q 3.1 are presented. The infant presented antimongoloid palpebral clefts, fine lips, bilateral microtia, deep eyes and microrretrognatia, among other dismorphies. The malformative picture included cataracts, microphthalmia, aorta coarctation and asymmetry of the brain lateral ventricles. It was undertaken a cytogenetic study, whose resulting chromosomal formula was 46, XY, dup (2) (q 2.1-q 3.1). The patient died at the age of 5 months due to a complicated pneumonia.
Key words: Structural chromosomal aberration, duplication 2q 2.1- q 3.1, partial trisomy 2q 2.1 - q 3.1.
INTRODUCCIÓN
El desarrollo de las técnicas de bandeo ha permitido determinar aproximadamente tres tipos de aberración por cada par cromosómico, dato que podría estar enormemente sesgado por las posibilidades de viabilidad de los cigotos. El resultado es alrededor de cien síndromes cromosómicos, unos mejor delineados que otros.1 Aun con el mejoramiento de las técnicas de que se dispone hoy día, es muy difícil el diagnóstico de las aberraciones de estructura.
Estas anomalías requieren mayor profundización para definir el cuadro malformativo y, sobre todo, su patrón dismórfico. Se presenta el caso siguiente con el objetivo de contribuir al mejor conocimiento del síndrome cromosómico producido por la duplicación parcial 2q 2.1-q 3.1.
PRESENTACIÓN DEL CASO
Lactante de 45 días, hijo de padres no consanguíneos, nacido a término por cesárea después de un embarazo complicado con hipertensión arterial materna. Presentó hipoxia al momento del parto y se mantuvo en observación sin necesidad de que se le administrara oxígeno ni antibióticos. Presentó hiperbilirrubinemia que fue tratada con fototerapia. Su peso al nacer fue de 4363 g y su talla de 50 cm, con una circunferencia cefálica de 35 cm.
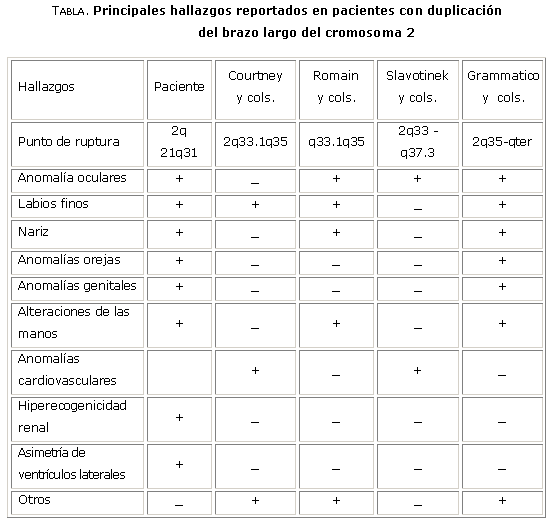
Al momento de su remisión al servicio de genética, el niño presentaba un patrón dismórfico caracterizado por ojos profundos y microftalmía, puente nasal alto con nariz algo elevada, desviación antimongoloide de las hendiduras palpebrales. Se encontró microtia bilateral con implantación baja de las orejas y hélix hiperenrollado. Los labios eran finos, con las comisuras hacia abajo y el filtro largo, con los pilares borrados. Entre otras malformaciones se hallaron catarata, hernia umbilical y en la región sacrocoxígea, un angioma plano y aumento de la coloración del vello a ese nivel. En el examen de los genitales se observó escrotalización del pene con criptorquidia derecha y testículo izquierdo retráctil. Las manos mostraban dedos largos y cabalgados, pulgares gruesos en «autostop» y surcos profundos en las palmas y también en las plantas (tabla).
En la radiografía de huesos largos se hallo ensanchamiento de los extremos dístales de ambos fémures y en la ecocardiografía se encontró coartación de la aorta, con estenosis moderada que requería tratamiento quirúrgico y agujero oval permeable. Los riñones presentaban aumento de la ecogenicidad sin otra alteración en la ultrasonografía (US) abdominal. Asimismo se detectó asimetría de los ventrículos laterales (derecho 6,2 mm y el izquierdo 2,8 mm), en el US transfontanelar.
Se realizó cariotipo usando la técnica convencional y el bandeo G. Se encontró una duplicación intersticial en el brazo largo del cromosoma 2. La fórmula cromosómica se describe como 46, XY, dup (2) (q 2.1- q 3.1). En la figura puede observarse la duplicación 2q hallada en nuestro paciente, el cual falleció a los 5 meses de edad como consecuencia de una de una neumonía complicada.
DISCUSIÓN
Al menos uno de cada 200 niños nacidos vivos tiene una aberración cromosómica y estas constituyen el 50 % de todos los abortos espontáneos. La delineación fenotípica de las anomalías cromosómicas parciales ha sido difícil por lo escaso de los reportes en la literatura, unido a la pobre supervivencia de los pacientes y la variedad de los puntos de ruptura. El fenotipo clásico descrito con hipotonía, hipertelorismo, nariz corta y hacia arriba, labio superior fino, micrognatia, orejas bajas y malformadas y criptorquidia de los reportes de duplicaciones parciales de las bandas 2(q3) concuerda con lo encontrado en nuestro paciente (tabla), aunque no podemos definir si se debe a una inserción de material cromosómico extra o a una duplicación de novo, ya que los padres no han podido ser estudiados.2-5
La magnitud del segmento involucrado obviamente tiene un efecto de dosis sobre el fenotipo del individuo y a este hecho pueden deberse las diferencias del caso con lo comunicado por otros autores2 (tabla). Un reporte de investigadores neozelandeses sobre una mujer de 22 años con una trisomía parcial (46, XX, dir dup (2)(q33.1-q35) de novo confirmado por FISH ( hibridación in situ con fluorescencia) describe un fenotipo similar.3 Los hallazgos incluyen epicanto, clinodactilia, escoliosis, puente nasal ancho y plano, labio superior fino, filtro largo y cuello corto. Aunque el segmento duplicado es más pequeño, se mantienen hallazgos comunes al parecer determinados por un punto crítico para la aparición de estos (tabla ).
Las afectaciones oculares, aunque no exactamente las mismas, han sido reportadas por Cotlier.4 No se detectó ectasias intestinales, ausencia de la última costilla ni las anomalías renales reportadas en la literatura.5 El paciente falleció tempranamente y quizá por ello no se encontraron anomalías del tracto piramidal u otras alteraciones neurológicas. En cuanto a la presencia de anomalías cardiovasculares, otros autores también reportan que no detallan el tipo de malformación hallada.5
Aunque el efecto de dosis está presente como ocurre en otras anomalías cromosómicas, es difícil hallar una relación directa con las malformaciones encontradas, lo cual pudiera deberse a que determinados locus juegan un rol en la regulación epigenética o en procesos importantes del desarrollo, teniendo en cuenta la alta densidad de genes homeóticos en la zona.6
Algunos de los trabajos revisados incluyen malformaciones como hipoplasia genital masculina, paladar hendido, estenosis del cardias, digitalización del pulgar, hernia hiatal e inguinal y malformaciones renales.5 Muchos de estos hallazgos son inespecíficos y frecuentes en las aberraciones cromosómicas, por lo que podrían reflejar un fenómeno general de interferencia en la morfogénesis como resultado del desequilibrio genético, más que la consecuencia de una idéntica trisomía parcial. Aunque el fenotipo facial para la trisomía parcial pura 2q3 ha sido descrito, se mantiene alguna controversia sobre las posibles bandas implicadas en las características faciales. Algunos autores refieren que hay evidencia suficiente para plantear que las bandas 2q33 a 2q37 son las responsables de la apariencia facial del síndrome.7-9
Por ser pocos los reportes de una aberración cromosómica similar y debido al solapamiento que puede haber en los puntos de ruptura, es difícil determinar si las diferencias en los hallazgos clínicos entre los casos previamente reportados y el nuestro se deben a los diferentes puntos de ruptura o a la variación clínica, relativamente frecuente en las anomalías cromosómicas.
REFERENCIAS BIBLIOGRÁFICAS
1. Mueller RF, Young ID. Chromosomal Aberrations. In: Emery"s. Elements of Medical Genetics 10 eds. Harcourt Health Sciences, 2001. Pp 246-64.
2. Kyllerman M, Wahlstrom J, Westerberg B, Gustavson KH. Delineation of a characteristic phenotypic in distal trisomy 2q. Helv Paediatr Acta. 1984dic;39(5-6):499-508.
3. Romain DR, Mackenzie NG, Moss D, Columbano-Green LM, Smythe RH. Partial trisomy for 2q in a patient with dir dup(2) (q33.1q35). J Med Genet. 1994 aug:31(8):52-3.
4. Cotlier E, Reinglass H, Rosenthal I. The eye in the partial trisomy 2q syndrome. Am J Ophthalmol. 1977; 84:251-8.
5. Schinzel A. Catalogue of unbalanced chromosome aberrations in man. Berlin- New York: Walter de Gruyter; 1984. Pp. 112-15.
6. Online Mendelian Inheritance in Man [database on the Internet]. Available at: http://www3.ncbi.nlm.nih.gov/omin
7. Grammatico P, Di Rosa C, Rinaldi R, Roccella M, Cupilari F. 2q35qter duplication syndrome: phenotypic definition. Genet Couns. 1997;8(4):327-34.
8. Drake Sebold C, Romie S, Szymanska J, Torres-Martinez W, Thurston V, Muesing C, et al. Partial trisomy 2q: Report of a patient with dup (2)(q33.1q35) . Am J Med. Genet A. 2005;134(1):80-3.
9. Slavotinek AM, Boles D, Lacbawan F. A female infant with duplication of chromosome 2q33 to 2q37.3. Clin Dysmorphol. 2003;12(4):251-6.
Recibido: 11 de octubre de 2007.
Aprobado: 26 de diciembre de 2007.
Lic. Marta M. Acosta Sabatés. San Francisco y Perla, Altahabana, La Habana, Cuba. Correo electrónico: martam.acosta@infomed.sld.cu


{kind=link}
{kind=link}