










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Pediatría
versión On-line ISSN 1561-3119
Rev Cubana Pediatr v.81 n.1 Ciudad de la Habana ene.-mar. 2009
Sospecha de enfermedad neuromuscular en el niño «asintomático» con aumento de la transaminasa glutámico-pirúvica o la creatina-fosfocinasa
Suspicion of neuromuscular disease in asymptomatic children with increase of glutamic-pyruvic transaminase or creatinine phosphokinase
Ramiro Jorge García García
Doctor en Ciencias Médicas. Profesor Titular. Hospital Pediátrico «Juan Manuel Márquez». La Habana, Cuba.
RESUMEN
El aumento prolongado de la transaminasa glutámico-pirúvica, sin causa demostrada de afectación hepática, o la elevación de los niveles de la creatina-cinasa en niños con pocos síntomas de trastorno neuromuscular o sin ellos, es motivo poco frecuente de consulta en neuropediatría. Se presenta un resumen de lo reportado al respecto en la literatura médica, y se incluyen comentarios al respecto y la propuesta de un algoritmo para el diagnóstico.
Palabras clave: Transaminasa glutámico-pirúvica, creatina-cinasa, enfermedad neuromuscular, hiperCKemia idiopática, distrofia muscular asintomática.
ABSTRACT
A lengthy increase of glutamic-pyruvic transaminase, without a proved cause, or rise of creatinine phosphokinase levels in children with a few symptoms of neuromuscular disorder or without them, is an infrequent reason of consultation in Neuropediatrics service. Authors present a summary of features reported in this respect in medical literature, and are included comments, as well as proposal of a algorithm for diagnosis.
Key words: Glutamic-pyruvic transaminase, creatinine phosphokinase, neuromuscular disease, idiopatic hyperkalemia, asymptomatic muscular dystrophy.
INTRODUCCIÓN
Cualquier trastorno que ocasione daño de las fibras musculares estriadas puede ser la causa de que las enzimas intracelulares pasen a la sangre, lo cual provoca un aumento variable de dicha concentración en sangre, que depende fundamentalmente del origen y la intensidad de la afectación. La determinación de la creatina-fosfocinasa o creatina-cinasa (CK, a partir de la denominación en inglés creatine kinase) es la evaluación enzimática más sensible para identificar el daño muscular; y las distrofias musculares (DM) son el motivo más frecuente de elevación importante de esta enzima en los niños con enfermedades neuromusculares.1
En la infancia, las enfermedades neuromusculares que se diagnostican con mayor frecuencia son las distrofias musculares (DM). Entre ellas, la de mayor presentación es la distrofia muscular de Duchenne (DMD) con una incidencia de 1 cada 3500 varones nacidos vivos, seguida por la Distrofia muscular de Becker (DMB); ambas son ligadas al sexo y se originan por deficiencias en la síntesis de la enzima de membrana conocida como distrofina (se denominan distrofinopatías). Esta deficiencia se debe a la mutación de un gen localizado en la región Xp 21 que es responsable de la codificación de esta proteína.2
Otras enfermedades neuromusculares en las que puede encontrarse el aumento enzimático (aunque en mucha menor cuantía) y que también se inician en la infancia son menos frecuentes que la DMD, como por ejemplo las atrofias musculares espinales (AME), con una incidencia de 7,8 - 10 casos x 100 000 nacidos vivos.3 En otras se presentan dificultades para realizar algunos exámenes «especiales» necesarios para el diagnóstico y por lo tanto puede existir una prevalencia mayor que la reportada, como ocurre con algunas miopatías congénitas o mitocondriales, y en otro grupo, el diagnóstico se retrasa por diversos motivos, como pueden ser las dificultades en la identificación de los síntomas cuando son ligeros, o cuando se acompañan de manifestaciones clínicas «muy evidentes y preocupantes» (crisis epilépticas o retraso mental), las que hacen más difícil el reconocimiento.4
En general, los pacientes que son remitidos a las consultas de Neuropediatría por una posible enfermedad neuromuscular, son enviados por presentar manifestaciones indiscutibles de este trastorno, como son la debilidad muscular, la disminución del tono o el trofismo muscular, de los reflejos osteotendinosos, o de la sensibilidad. 5,6.
Sin embargo, no siempre las características clínicas son tan evidentes y en ocasiones son consultados inicialmente por otros especialistas, quienes consideran posteriormente remitir al paciente al neuropediatra, para definir si el motivo es una enfermedad del nervio periférico (neuropatía) o del músculo (miopatía). Aquí se incluyen los niños con trastornos de la marcha, deformidades esqueléticas, caídas frecuentes, dudosa debilidad muscular y aumento de la transaminasa glutámico-pirúvica (TGP), la transaminasa glutámico-oxalacética (TGO) y la creatina-cinasa (CK), detectados «accidentalmente».7 Aquellos con pocas o ninguna manifestación clínica, pero con aumento de los niveles enzimáticos, constituyen posiblemente el mayor reto para el diagnóstico.
Son pocos los reportes con relación al aumento de la de la TGP o la CK, en niños en los que se presenta poca o ninguna sintomatología que justifique la elevación de los niveles de estas enzimas, pero el neuropediatra debe determinar principalmente la presencia de un trastorno neuromuscular. Lo anterior es el motivo de este trabajo, donde se analiza lo reportado en la literatura médica y se emiten comentarios que incluyen una propuesta de algoritmo para el diagnóstico.
Se revisaron textos básicos de Neurología y Pediatría, para resumir los elementos clínicos fundamentales de las enfermedades neuromusculares, los motivos de las variaciones en los niveles enzimáticos y los métodos de diagnóstico.
Se realizó una búsqueda en PubMed/Medline, según recomendaciones publicadas para alcanzar mayor efectividad.8 Se seleccionaron los límites siguientes: con resumen, publicación en los últimos 10 años, artículos de revisión; idioma inglés, francés o español. En los seleccionados, se solicitó el texto completo y fueron excluidos aquellos en que no fue posible la obtención del artículo.
EL NIÑO CON AUMENTO DE LA TRANSAMINASA GLUTÁMICO-PIRÚVICA
En la consulta de neuropediatría se reciben niños sobre quienes inicialmente no se pensó en la posibilidad de que presentaran una enfermedad neuromuscular, a los que se les realizó la determinación de TGP por diferentes causas y esta arrojó resultados elevados. Lógicamente, ante estos casos, el médico de asistencia piensa en primer lugar en el diagnóstico de hepatitis de posible origen viral y se inicia un largo proceso para confirmar esta impresión e intentar determinar su origen.9 Luego de un período mayor o menor se llega a la conclusión de que el aumento se debe posiblemente a una enfermedad neuromuscular y entonces se remite el niño a la consulta de neuropediatría.
Cuando estos pacientes llegan a la consulta, el primer paso es la realización de un interrogatorio exhaustivo para detectar fundamentalmente las manifestaciones clínicas que sugieran una miopatía y, posteriormente, el examen físico para determinar la existencia de signos que justifiquen el diagnóstico.6 A veces, desde la primera consulta se comprueba una debilidad muscular generalmente proximal, con disminución o pérdida de los reflejos osteotendinosos y disminución del trofismo de igual topografía, lo que apoya la posibilidad de una miopatía.6 En otros casos solamente se encuentran manifestaciones ligeras o ninguna manifestación de afectación muscular. O sea, el diagnóstico puede ser evidente, o por el contrario resultar un reto a la capacidad diagnóstica.
En ambos casos se hace necesario inicialmente realizar otras determinaciones enzimáticas (transaminasas, CK, aldolasa y LDH), estudios neurofisiológicos (electromiografía [EMG] y los estudios de conducción nerviosa [ECN], fundamentalmente) y se debe tener en cuenta la necesidad de realizar una biopsia muscular y estudios genéticos.1
En los pacientes en que se encuentra también la CK elevada, el diagnóstico de DM es muy probable y la realización del EMG puede presentar un patrón miopático,1 lo que apoya aún más el diagnóstico e incluso puede ser suficiente para diferir la realización de la biopsia muscular. Lo más frecuente es que se trate de un varón y los estudios de deleción (eliminación) del exón presenten alteraciones que indiquen la existencia de una DM por una mutación genética ligada al cromosoma X.2
En los 17 años de trabajo del Servicio de Neuropediatría del Hospital Pediátrico «Juan Manuel Márquez», se han recibido 4 pacientes atendidos previamente por hepatitis de probable origen viral y evolución prolongada, a los que después de concluidos los estudios se determinó que requerían ser consultados por el neuropediatra, por la posibilidad de que los cambios estuvieran relacionados con una enfermedad neuromuscular (datos no publicados).
En tres de ellos, se recogió el antecedente de caídas frecuentes y dificultades para subir escaleras. En los cuatro niños se encontró aumento de la CK y en tres se comprobó por EMG y biopsia el diagnóstico de DMD. En el otro paciente (asintomático), el EMG fue normal, pero el estudio genético comprobó la deleción del exón compatible con DM y valores de CK por encima de 9000 UI/L, por lo que se planteó el diagnóstico de distrofinopatía.
EL NIÑO CON AUMENTO DE LA CREATINA-CINASA
En la práctica, los aumentos de la CK se encuentran con mayor frecuencia en las miopatías y, por tanto, no es sorprendente que se encuentren en niños con manifestaciones evidentes de enfermedad muscular, sobre todo las DM, pero en los niños asintomáticos o con síntomas ligeros se hace necesario precisar su origen real.10
Ocasionalmente se pueden encontrar pacientes con aumento de la CK en todas las edades y las causas más comunes son los infartos, trastornos musculares, el ejercicio exagerado y la medicación con drogas para disminuir el colesterol. Con menor frecuencia se puede encontrar en el embarazo, la hipertermia y en las anomalías en tiroides y paratiroides, y es importante tener en cuenta que pueden existir niveles altos de CK en pacientes con rabdomiólisis.11,12
El aumento de la CK puede ser un signo de trastorno neuromuscular subclínico en pacientes sin anomalías neurológicas manifiestas, e incluso el valor de la biopsia y los estudios EMG pueden ser de poca utilidad, excepto en aquellos con distrofinopatías. Se reporta la utilidad de la biopsia muscular en pacientes en los que se encuentra ocasionalmente un aumento de la CK sin manifestaciones clínicas, sobre todo si se encuentran valores que excedan cinco veces el valor máximo normal.13
En un estudio realizado por Dabky y colaboradores (2006), se encontraron estas características en aproximadamente la mitad de los casos, donde la biopsia muscular fue anormal y se encontraron resultados específicos de distrofia muscular en el 8 % de los casos.10
En otra investigación realizada por Fernandez y colaboradores, en la que se incluyen los resultados de las biopsias musculares realizadas a 104 pacientes con aumento de la CK (>500 UI/L), sin signos de debilidad muscular, los autores pudieron determinar diagnósticos a partir de ésta en el 55 % de los casos e identificaron a las enfermedades por acumulación de glucógeno como las de mayor frecuencia, seguidas por las distrofias musculares y las miopatías inflamatorias. Concluyeron con la afirmación de que la mayor probabilidad de diagnóstico por biopsia se encuentra en los niños con aumento de la CK por encima de 2000 UI/L.14
En este estudio se incluyeron pacientes adultos, lo que influye en la distribución de las causas de los aumentos enzimáticos. La afirmación sobre el valor de la biopsia en niños con aumento de la CK es válida en pediatría, ya que constituye uno de los pilares del diagnóstico en los trastornos neuromusculares, muchos de ellos difíciles de precisar. Si embargo, según muchos autores, en aquellos casos en que las manifestaciones clínicas son claras y se presentan alteraciones en los valores de las enzimas y la neurofisiología, sobre todo si pueden realizarse los estudios genéticos, se puede valorar diferir su realización.
En la infancia es importante considerar siempre el diagnóstico de enfermedades neuromusculares, incluso subclínicas. Melis y colaboradores (1998) reportaron tres niños asintomáticos, de tres familias diferentes, a quienes se diagnóstico una distrofinopatía y en los estudios específicos se encontró deleción del exón de variable extensión en el gen relacionado con la distrofina. Al estudiar a los familiares, hallaron la misma deleción en adultos asintomáticos (o con ligeros síntomas) y niveles de CK normales pero cercanos al límite superior de la normalidad.15
Por su parte, Saengpattrachai y otros (2006) reportaron tres varones (de 7, 8 y 67 años), de una misma familia, con diagnóstico de distrofinopatía asintomática. En estos casos se comprobó la deleción del exón, y en los análisis genéticos se identificó igual deleción en cuatro hembras, a las que consideraron portadoras.16
En los casos en que se descartan las causas conocidas de aumento de las CK y las enfermedades neuromusculares mediante la clínica, neurofisiología y la biopsia muscular, se puede plantear el diagnóstico de aumento idiopático de la CK (hiperCKemia idiopática), trastorno descrito desde 1980.11
De lo anterior se puede concluir que es muy importante sospechar la presencia de una enfermedad neuromuscular en los niños con aumento de TGP por tiempo prolongado y sin comprobación del origen de la afectación hepática, incluso en pacientes sin síntomas de trastorno neuromuscular. En esos casos, es muy importante la determinación de otras enzimas (CK y TGO) y en el caso en que se encuentre elevada la CK, es muy probable el diagnóstico de enfermedad neuromuscular y sobre todo de DM, por lo que se imponen los estudios neurofisiológicos.
La ejecución de una biopsia muscular debe considerarse de acuerdo con las opciones de realizar estudios genéticos, y la determinación de las deleciones y de la distrofina, sin olvidar la posibilidad de DM asintomática y de la hiperCKemia idiopática.
¿Cuáles son las características del aumento idiopático de la creatina-cinasa?
El aumento persistente de la CK puede encontrarse ocasionalmente en las personas sanas. En 1980 Rowland nombró este trastorno hiperCKemia idiopática.17
En 1988, Brewster y Visser reportan 14 pacientes con aumento de la CK sin causa demostrable, incluidas las enfermedades neuromusculares.18 Más tarde, D' Adda y colaboradores reportaron en el 2006 los resultados de un estudio de seguimiento de pacientes con aumento de la CK sin síntomas acompañantes o con poca sintomatología, y pudieron precisar la causa del aumento en 21/114 (18 %). Cincuenta y cinco de los 93 restantes fueron seguidos por 6 años y la mayoría siguió asintomática; en casi todos disminuyeron los valores de la CK y en 12 se normalizaron evolutivamente.19
PROPUESTA DE ALGORITMO PARA EL DIAGNÓSTICO DE PACIENTES CON AUMENTOS DE LA TGP Y LA CK
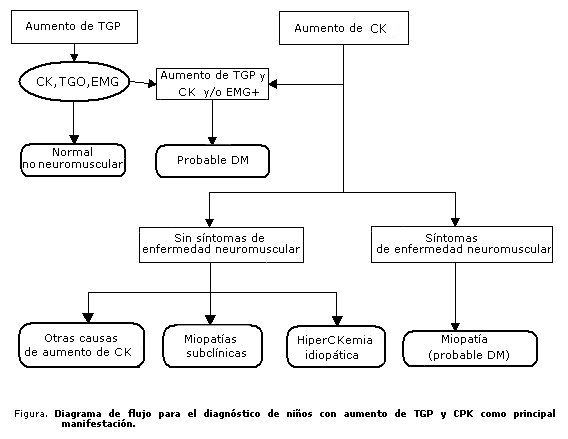
Conclusiones
En niños con aumentos de la TGP sin causa comprobada de enfermedad hepática y evolución prolongada debe tenerse en cuenta la posibilidad de enfermedad neuromuscular, incluso en ausencia de manifestaciones clínicas. En los pacientes en que se encuentra asociada la elevación de la CK, el diagnóstico de estos trastornos es más probable y, principalmente, las distrofias musculares.
En aquellos con aumento de la CK deben descartarse un grupo de enfermedades que pueden ser las causales, pero debe pensarse sobre todo en las enfermedades neuromusculares y realizar los estudios adecuados de acuerdo con las características de cada paciente, teniendo en cuenta la posibilidad diagnóstica de DM asintomática. En el caso en que no pueda confirmarse el origen de los valores elevados de CK se puede hacer el diagnóstico de hiperCKemia idiopática.
REFERENCIAS BIBLIOGRÁFICAS
1. Adams RD, Victor M. Métodos auxiliares de laboratorio en el diagnóstico de la enfermedad muscular. En: Adams RD, Victor M. Principios de Neurología. La Habana: Edición Revolucionaria; 1982. Pp. 982-1001.
2. Twee D. Muscular Dystrophy. [monograph on internet] Available from: http://www.emedicine.com/orthoped/TOPIC418.HTM Consultado: 3 marzo, 2007.
3. Tsao B. Spinal Muscular Atrophy. [monograph on internet] Available from: http://www.emedicine.com/neuro/TOPIC631.HTM Consultado: 2 de nov, 2006.
4. Colomer J, Iturriaga C. Patología neuromuscular. En: Neurología Pediátrica. Madrid: Ediciones Ergon; 2000. Pp. 425-65.
5. Adams RD, Victor M. Enfermedades de los nervios periféricos. En: Adams RD, Victor M. Principios de Neurología. La Habana: Edición Revolucionaria; 1982. Pp. 453-508.
6. Adams RD, Victor M. Principios de la miología clínica. Clasificación sindrómica de las enfermedades musculares. En: Adams RD, Victor M. Principios de Neurología. La Habana: Edición Revolucionaria; 1982. Pp. 971-81.
7. Kleppe B, Reimers CD, Altmann C, Pangratz DE. Findings in 100 patients with idiopathic increase in serum creatin kinase activity. Med Klin 1999; 90(11):623-7.
8. Barroso Espadero D, Orejón de Luna G, Fernandez Rodriguez M. Introducción a MEDLINE y las búsquedas bibliográficas II. Guía de uso de PubMed. Rev Pediatr Aten Primaria 2004;6:77-112.
9. Brines J, Codoñer P. Hepatopatías agudas. Hepatitis vírica. En: Cruz M, editor. Tratado de Pediatría. 7a edición. La Habana: ECIMED; 2006. Pp.1191-207.
10. Dabky R, Sadeh M, Herman O, Berger E, Watenberg N, Hayek S, et al. Asymptomatic or minimally symptomatic hyperCPKemia: histopathologic correlates. Isr Med Assoc J 2006;8(2):110-3.
11. Douglas K. HyperCPKemia: a diagnostic dilemmas. Can Fam Physician 2005;51(2):240-1.
12. Morandi L, Angelini C, Prelle A, Pini A, Grassi B, Bernardi G, et al. High plasma creatine kinase: review of the literature and proposal for a diagnostic algorithm. Neurol Sci 2006;27:303-11.
13. Wokke JH. Raised creatine - kinase serum activity: not neccesarily a sign of disease. Ned Tijdschr Geneeskd 2003;147(41):1988-2000.
14. Fernandez C, De Paula AM, Figarella-Bronger D, Krahn M, Giorgi R, Chabrol B, et al. Diagnostic evaluation of clinically normal subjects with chronic hyperCPKemia. Neurology 2006;66(10):1585-7.
15. Melis MA, Cau M, Muntoni F, Mateddu A, Galanello R, Boccone L, et al. Elevation of serum creatine kinase as the only manifestation of an intragenic deletion of the dystrophin gene in three unrelated families. Eur J Paediatr Neurol 1998;2(5):255-61.
16. Saengpattachai M, Ray PN, Howkins CE, Berzen A, Bonwell BL. Grandpa and I have dystrophinopathy?: approach to asymptomatic hyperCPKemia. Pediatr Neurol 2006;35(2):145-9.
17. Prelle A, Tancredi L, Sciacco M, Chivero L, Battisted A, Bazzi P, et al. Retrospective study of a large population of patients with asymptomatic or minimally symptomatic raised serum creatinine kinase levels. J Neurol 2002;249(3):305-11.
18. Brewster LM, de Visser M. Persistent hyperCPKemia: fourteen patients studied in retrospect. Acta Neurol Scand 1988;77(1):60-3.
19. D'Adda E, Sciacco M, Freeguglietti ME, Crugnola V, Lucchini V, Martinelli-Boneschi F. Follow-up of a large population of asymptomatic/oligosymptomatic hiperCPKemic subjects. J Neurol 2006; 253(11):1399-1403.
Recibido: 12 de junio de 2008.
Aprobado: 26 de septiembre de 2008.
Ramiro Jorge García García. Hospital Pediátrico «Juan Manuel Márquez». Avenida 31 y 76, Marianao. La Habana, Cuba.
Correo electrónico: ramirogg@infomed.sld.cu

