










Meu SciELO
 Serviços customizados
Serviços customizadosServiços Personalizados
Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkRevista Cubana de Pediatría
versão On-line ISSN 1561-3119
Rev Cubana Pediatr v.81 n.4 Ciudad de la Habana set.-dez. 2009
Infartos cerebrales de repetición y anemia drepanocítica en un niño: revisión de la literatura médica
Repeated brain infarctions and sickle cell anemia in a child: medical literature review
José Vargas Díaz,I Reinaldo Puga Gómez,II Jorge Luis Seijo Hernández,III Luis Quevedo Sotolongo,IV Patricia Isabel Corona Rodríguez,V Antonia Izaguirre Corrales VI
I Especialista de II Grado en Pediatría y Neurología. Máster en Atención Integral al Niño. Profesor Titular de Pediatría. Investigador Auxiliar. Universidad Médica de la Habana. Cuba.
II Especialista de II Grado. Profesor Auxiliar de Pediatría. Universidad Médica de la Habana. Clínica Central «Cira García». La Habana, Cuba.
III Especialista de I Grado en Pediatría. Instructor. Clínica Central «Cira García». La Habana, Cuba.
IV Profesor Auxiliar de Radiología. Universidad Médica de la Habana. Clínica Central «Cira García». La Habana, Cuba.
V Especialista de I Grado en Hematología. Instructora. Universidad Médica de La Habana. Cuba.
VI Especialista de I Grado en Pediatría. Clínica Central «Cira García». La Habana, Cuba.
RESUMEN
Una de las complicaciones neurológicas más devastadoras de la anemia drepanocítica son los ictus, tanto isquémicos como hemorrágicos. El 11% de los pacientes con hemoglobina SS (HbSS) tienen un ictus antes de los 20 años de edad. Se presenta el caso de un niño de 14 años, congolés, gravemente desnutrido, con anemia drepanocítica y antecedentes de ictus isquémicos de repetición, que fue atendido en la Clínica Internacional «Cira García». La resonancia magnética evidenció signos de infartos antiguos a diferentes niveles en ambos hemisferios y zonas de encefalomalacia. Este paciente muestra la evolución natural de las complicaciones cerebrovasculares de la anemia de células falciformes.
Palabras clave: Infartos cerebrales, ictus, anemia drepanocítica, hidroxiurea, ultrasonido Doppler transcraneal.
ABSTRACT
Among the most devastating neurologic complications from sickle-cell anemia are the ischemic and hemorrhagic ictus. The 11% of patients with SS hemoglobin (HbSS) has ictus before the twenties. This is the case of a child from the Congo aged 14 severely undernourished presenting with sickle-cell anemia and backgrounds of repeated ischemic ictus, seen in the "Cira García" International Clinic. Magnetic resonance showed signs of an old infarction at different levels of both hemispheres and encephalomalacia zones. This patient shows the natural course of the cerebrovascular complications of sickle-cell anemia.
Key words: Brain infarctions, ictus, sickle-cell anemia, hydroxyurea, transcranial Doppler US.
INTRODUCCIÓN
En los últimos años ha habido un interés creciente por la fisiopatología de la anemia drepanocítica (AD) y sus complicaciones en la infancia.1 Actualmente los accidentes cerebrovasculares (ictus) en los niños se diagnostican con más frecuencia porque se piensa en ellos y por disponer de técnicas de imágenes con gran capacidad de resolución y menos riesgosas para los niños.
La incidencia de los ictus en los niños menores de 15 años se ha reportado en un amplio rango, desde 2,5 a 13 por 100 000 por año.2-4
Los ictus son una de las complicaciones mayores de la AD.4,5 La frecuencia de los ictus en los niños con AD es mayor que en el resto de los niños. Se ha reportado una incidencia de 285 por 100 000 por año, tanto para los ictus isquémicos como para los hemorrágicos. En niños con AD la incidencia por 100 000 pacientes con AD fue de 238 para los ictus isquémicos y de 47,5 para los hemorrágicos.
La mayor incidencia para el primer ictus en niños con AD se observa en dos picos, entre los 2 a 5 años y luego de 6 a 9 años, con incidencias de 1,02 % y del 0,68 %, respectivamente.6-8
Los pacientes con AD pueden expresar el compromiso cerebrovascular de diferentes formas, desde las asintomáticas con comprobación de infarto cerebral, mediante resonancia magnética nuclear (RMN), hasta las formas sintomáticas cuya clínica depende del tamaño y localización de la lesión.
Desde el punto de vista del territorio vascular más afectado, la porción supraclinoidea de la arteria carótida interna (ACI) y la arteria cerebral media son las más afectadas y en menor proporción la arteria cerebral anterior y las arterias del territorio posterior. Algunos pacientes presentan una vasculopatía progresiva de la ACI con el desarrollo de vasos colaterales o síndrome de Moyamoya.9,10
Los niños con AD pueden presentar trombosis senovenosas cerebrales o incluso síndrome de la arteria espinal anterior. La hemorragia subaracnoidea y la hemorragia intracraneal pueden ocurrir en el contexto de la trombosis senovenosas y después de rupturas de aneurismas. En estos niños también se
ha descrito la leucoencefalopatía posterior reversible luego de un síndrome torácico agudo,11,12 pero puede también resultar en un infarto occipital.
Entre los factores de riesgo (FR) para un primer ictus en un paciente con AD se han reportado los siguientes: velocidades del flujo sanguíneo elevadas en el ultrasonido Doppler transcraneal (UDT), niveles bajos de hemoglobina, conteo de leucocitos elevados, hipertensión arterial, historia de infartos silentes y antecedentes de crisis torácicas.13,14
La anemia hemolítica y la oclusión vascular se sobreponen en la propensión a padecer los subfenotipos de hipertensión pulmonar, accidente vascular encefálico, priapismo y úlcera maleolar.1
La oclusión vascular es favorecida por la generación de trombina in vivo. El endotelio activado y la fosfatidilserina en la superficie de los hematíes condicionan un estado trombofílico.1 El aumento del fragmento 1,2 de la protrombina y el aumento de la fosfatidilserina del glóbulo rojo se correlacionan con el aumento de la velocidad del flujo sanguíneo medido por ultrasonido Doppler transcraneal en algunas arterias cerebrales.1
El identificar por UDT a estos pacientes en riesgo alto de un ictus brinda una oportunidad de prevenirlos.15-17 Los niños que presentan velocidades del flujo sanguíneo igual o superior a 200 cm/s tienen un riesgo de ictus de al menos un 10 % por año.18,19
Existe una predisposición familiar a ictus en pacientes con AD, y se han identificado identificado factores genéticos20 y otros no genéticos como la pobre nutrición y la polución ambiental. La hipoxemia nocturna en estos niños puede ser un factor de riesgo modificable para enfermedades del sistema nervioso central e ictus.21
El riesgo de recurrencia en AD es alto. En algunas series se ha reportado un 23 % de recurrencia para una primera recurrencia a los 4 años de evolución y en general un promedio de recurrencia de 2,2 por 100 pacientes por año.22 Dobson y cols.23 reportan un 41 % de pacientes con ictus recurrentes o accidentes transitorios isquémicos (ATI). La recurrencia resultó más común en los pacientes que presentaron un síndrome de Moyamoya.
Los pacientes con AD pueden presentar lesiones cerebrales subclínicas llamadas silentes (20 %), detectadas por la RMN cerebral, las cuales predominan en las regiones corticales frontales y parietales, así como en zonas subcorticales y en regiones llamadas fronteras.24 Estos infartos «silentes» son causa importante de deterioro de las funciones cognitivas, que se expresan como trastornos del aprendizaje y de la conducta.25,26
Los niños con AD pueden presentar todas las modalidades de hemorragias intracraneales.27,28 La asociación de síndrome de Moyamoya, de trombosis senovenosa, de hipertensión arterial, uso de esteroides o de transfusión de sangre reciente, incrementan el riesgo para hemorragias intracraneales en estos enfermos.
El tratamiento de pacientes con AD, si se trata de un ictus agudo, consiste en hidratación y exanguinotransfusión.29,30 La exanguinotransfusión evitaría el riesgo de incremento de la viscosidad sanguínea, que podría acompañarse de un rápido incremento del hematocrito (clase IIa; nivel de evidencia C). Se debe evitar la hipoxemia e hipotensión y mantener al paciente normoglicémico. De igual modo, es importante insistir en identificar y de ser así tratar, la infección, el cardioembolismo y la trombosis senovenosa.31
Es muy importante la prevención de un ictus, tanto primaria como secundaria, en pacientes con AD. El uso de transfusiones periódicas de sangre, la hidroxiurea y el trasplante de médula ósea constituyen modalidades de tratamiento con diversos grados de evidencia, así como en pacientes con Moyamoya lo constituyen el uso de procederes de anastomosis, del tipo de la encefaloduroarteriosinangiosis.32-36
Las transfusiones periódicas dirigidas a reducir el porcentaje de hemoglobina S son efectivas para disminuir el riego de ictus en niños entre 2 y 16 años de edad, que presentan un resultado anormal en el UDT (nivel de evidencia A).
En niños con AD, en quienes se haya confirmado un infarto cerebral, deben incluirse un programa regular de transfusión de glóbulos rojos y de medidas de prevención del depósito de hierro (clase I, nivel de evidencia B).37 La hidroxiurea ha sido usada para disminuir los episodios dolorosos en adultos con AD y aún no se dispone de evidencias de si reduce el riego de ictus en niños con AD. Se ha postulado que la disminución del óxido nítrico y el aumento de la endotelina-1 desempeñan un papel esencial en el tono vasomotor y en las alteraciones del endotelio. El óxido nítrico es un potente vasodilatador y regula la adherencia de las células, la agregación plaquetaria y la producción de eicosanoides como la prostaciclina. La endotelina-1 es un potente vasoconstrictor y es proinflamatoria. La hidroxiurea disminuye la concentración de endotelina-1.1
La evidencia de que se dispone recomienda que la hidroxiurea puede ser considerada en niños con AD e ictus, cuando el programa de transfusiones crónicas debe ser interrumpido (clase IIb, nivel de evidencia B).38
Los procederes quirúrgicos de revascularización podrán ser considerados en pacientes con AD quienes continúen presentando disfunciones cerebrovasculares a pesar del manejo médico óptimo (clase IIb, nivel de evidencia C). El trasplante de médula ósea puede ser considerado en niños con AD (clase IIb, nivel de evidencia C).
PRESENTACIÓN DEL CASO
Escolar de trece años de edad, procedente del Congo, con antecedentes de anemia drepanocítica, que estuvo presentando episodios de síndrome mano-pie desde muy pequeño. A los 9 años de edad, presentó un episodio agudo, que fue considerado por los profesionales que lo atendieron como un ictus arterial isquémico. Antes hubo pérdida de habilidades motoras, del lenguaje, así como de otras funciones cognitivas, según recuerda el padre. Desde entonces el paciente se encuentra en sillón de ruedas y no puede deambular por sí solo.
Antecedentes patológicos personales:
- Paludismo (en 4 ocasiones).
- Parasitismo intestinal.
Antecedentes patológicos familiares:
- Tías maternas: asma bronquial.
- Abuela materna y tía materna: hipertensión arterial.
- Padre y madre: hemoglobinopatia AS (rasgo).
- Abuelo paterno: diabetes mellitus.
Datos positivos al examen físico:
- Coloración ictérica de piel y mucosas, incluida la conjuntiva ocular.
- Prognatismo maxilar superior. Disminución marcada del panículo adiposo con hipotrofia muscular generalizada y afectación nutricional en la línea del marasmo.
- Tiene sostén de cabeza, no cambia de decúbito por sí solo, no hay equilibrio de tronco, se mantiene sentado con apoyo. No se sostiene solo de pie. La marcha con apoyo del padre es muy rudimentaria.
- Afectación de la motilidad activa y pasiva, con limitación muy marcada de la articulación del codo derecho. Hiperreflexia osteotendinosa con clonus casi inagotable (bilateral), así como Babinski bilateral e hipertonía de los cuatro miembros. No hay afectación de pares craneales. Al interrogatorio evidencia afectación del lenguaje y de la memoria reciente y pasada.
- Hepatomegalia de 2 a 3 cm que rebasa el reborde costal derecho, bazo no palpable. Soplo sistólico II/VI en el borde esternal izquierdo.
Resultado de las investigaciones:
- Hemograma con anemia moderada y reticulocitosis.
- Alteración de las enzimas hepáticas.
- Coagulograma, glicemia y función renal dentro de parámetros normales.
- Electroencefalograma dentro de límites normales.
- Angiorresonancia de cráneo: atrofia cerebelosa, periventricular y cortical. Áreas de atrofia posinfarto en región frontal derecha y temporoparietal izquierda. Oclusión de ambas carótidas internas con gran desarrollo del sistema arterial posterior. Importantes áreas de encefalomalacia (figuras 1, 2 y 3).
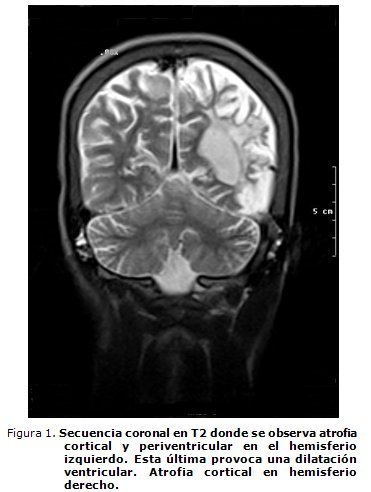
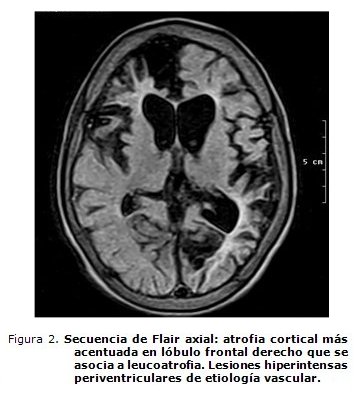

- Radiografía de pelvis ósea con parámetros aceptables para su enfermedad de base.
- Ultrasonido de hemiabdomen superior: vesícula de paredes engrosadas y bilis de estasis con múltiples cálculos pequeños en su interior, el mayor de 0,6 cm.
Evolución:
Sin complicaciones para su enfermedad de base, fue evaluado por especialistas de pediatría, ortopedia, medicina física y rehabilitación, nutrición hematología y neuropediatría.
Tratamiento adoptado:
- Dieta con incremento progresivo de energía y nutrientes.
- Hidroxiurea (500 mg): 1 cápsula diaria a las 8 a.m. (60 dosis).
- Neurovitán: Un vial bebible en almuerzo y comida.
- Ácido fólico: (1 mg): Una tableta diaria.
- Trofin: Dar 15 mL lunes y viernes.
- Eritropoyetina (bulbo 4000 U): Poner 1 bulbo subcutáneo 2 veces por semana (lunes y viernes), por 6 dosis.
- Omega 3: 500 mg al día.
Diagnóstico al egreso:
- Anemia drepanocítica.
- Cuadriparesia espástica secundaria a infartos cerebrales de repetición.
- Déficit intelectual.
- Desnutrición energética nutricional en la línea del marasmo.
- Litiasis biliar.
Recomendaciones:
- Continuar tratamiento médico, incluyendo dieta con incremento progresivo de la energía, hasta satisfacer sus necesidades totales con una proporción adecuada de macronutrientes que garantice una ingesta con una proporción de 150 cal/g de nitrógeno proteico aportado.
- Ingerir abundantes líquidos y evitar infecciones.
- Se recomendó al regreso a su país recibir atención especializada por hematología e iniciar régimen de trasfusiones sanguíneas cada 3 o 4 semanas o en su defecto tratamiento con hidroxiurea.
- Iniciar fisioterapia y rehabilitación.
- Comenzar educación especial.
DISCUSIÓN
Se trata de un adolescente congolés diagnosticado de padecer de anemia drepanocítica y que tenía antecedentes de episodios de síndrome «manos-pies». A los 5 años de edad presentó un evento neurológico agudo en su país, el cual fue catalogado como un ictus arterial isquémico. Perdió habilidades motoras y del lenguaje, y no volvió a deambular por sí solo desde entonces. Es conocido que los pacientes con AD hacen con frecuencia este tipo de eventos vasculocerebrales,6-8 y que éstos pueden ser además recurrentes.22,23
Los autores consideran que este paciente ha tenido eventos isquémicos de repetición, alguno de ellos posiblemente silente, quizás responsables del deterioro del aprendizaje y de la conducta que presenta y de las áreas de atrofia posinfarto en regiones frontal derecha y temporoparietal izquierdas, así como de las lesiones de encefalomalacia que evidencia la RMN cerebral realizada.25,26
La AD dejada evolucionar espontáneamente, se sabe, puede desarrollar una vasculopatía intracraneal progresiva y llegar ésta a un síndrome de Moyamoya.32-36 Este paciente tiene el contexto clínico e imagenológico para sospechar un síndrome de Moyamoya secundario a la AD, a pesar de que no se pudo opacificar el territorio carotídeo bilateralmente en la angio-RMN cerebral, por oclusión de ambas carótidas internas (figuras 1, 2 y 3).
El ultrasonido Doppler transcraneal en este niño no evidenció flujo sanguíneo a través de las carótidas intracraneales ni a nivel de las cerebrales medias y anteriores, expresión de la vasculopatía obstructiva que ya presenta.
Existen evidencias que indican que a todo niño con AD se le debe realizar un UDT, al menos una vez por año, en busca de un flujo sanguíneo elevado (mayor de 200 cm/s) que traduce un riesgo de ictus en el paciente. A este niño no se le realizó tal estudio, que pudo haber sido un indicador para tomar medidas que evitaran la evolución deteriorante que sufrió.15-17
El paciente no fue seguido en consulta y no recibió atención especializada para su anemia hemolítica, por lo que estuvo privado de los métodos en uso para mantener la concentración de la hemoglobina S por debajo de un 30 % y así evitar tanto el ictus primario como la recurrencia de éstos.31,38
La desnutrición proteico-energética que presenta el paciente es el resultado del gran catabolismo que ha experimentado por su enfermedad hemolítica crónica desatendida, la pobre ingesta de nutrientes en general y las infecciones repetidas.
Revertir esta tórpida evolución va ha ser tarea difícil, aunque mejorando la nutrición, evitando las infecciones y controlando los niveles de hemoglobina SS, bien con un régimen de transfusiones crónicas o en su defecto con el tratamiento con hidroxiurea, podría evitar nuevas complicaciones y hasta la muerte prematura de este niño.33-38
REFERENCIAS BIBLIOGRÁFICAS
1. Svarch E. Fisiopatología de la drepanocitosis. [monografía en Internet]. Disponible en: http://bvs.sld.cu/revistas//hih/vol25_1_09/hih03109.htm
2. Lieberman L, Kirby M, Ozolins L, Mosko J, Friedman J. Initial presentation of unscreened children with sickle cell disease: The Toronto experience. Pediatr Blood Cancer. 2009 Sep;53(3):397-400.
3. Schoenberg BS, Mellinger JF, Schoenberg DG. Cerebrovascular disease in infants and children: a study of incidence, clinical features, and survival. Neurology. 1978;28:7638.
4. Lynch JK, Hirtz DG, DeVeber G, Nelson KB. Report of the National Institute of Neurological Disorders and Stroke workshop on perinatal and childhood stroke. Pediatrics. 2002;109:11623.
5. Fullerton HJ, Adams RJ, Zhao S, Johnston SC. Declining stroke rates in Californian children with sickle cell disease. Blood. 2004;104:3369.
6. Prengler M, Pavlakis SG, Prohovnik I, Adams RJ. Sickle cell disease: the neurological complications. Ann Neurol. 2002;51:54352.
7. Meremikwu MM. Sickle cell disease. [web page] Clin Evid. 2009 Mar 27; available at: http://clinicalevidence.bmj.com/ceweb/conditions/bly/2402/2402_references.jsp
8. Earley CJ, Kittner SJ, Feeser BR, Gardner J, Epstein A, Wozniak MA, et al. Stroke in children and sickle-cell disease: Baltimore-Washington Cooperative Young Stroke Study. Neurology. 1998;51:16976.
9. Ohene-Frempong K, Weiner SJ, Sleeper LA, Miller ST, Embury S, Moohr JW, et al. Cerebrovascular accidents in sickle cell disease: rates and risk factors. Blood. 1998;91:28894.
10. Amlie-Lefond C, Bernard TJ, Sébire G, Friedman NR, Heyer GL, Lerner NB, et al.; International Pediatric Stroke Study Group. Predictors of cerebral arteriopathy in children with arterial ischemic stroke: results of the International Pediatric Stroke Study. Circulation. 2009;119(10):1361-2.
11. Khademian Z, Speller-Brown B, Nouraie SM, Minniti CP. Reversible posterior leuko-encephalopathy in children with sickle cell disease. Pediatr Blood Cancer. 2009 Mar;52(3):373-5.
12. Henderson JN, Noetzel MJ, McKinstry RC, White DA, Armstrong M, DeBaun MR. Reversible posterior leukoencephalopathy syndrome and silent cerebral infarcts are associated with severe acute chest syndrome in children with sickle cell disease. Blood. 2003;101:4159.
13. Armstrong-Wells J, Grimes B, Sidney S, Kronish D, Shiboski SC, Adams RJ, et al. Utilization of TCD screening for primary stroke prevention in children with sickle cell disease. Neurology. 2009;72(15):1316-21.
14. Ohene-Frempong K, Weiner SJ, Sleeper LA, Miller ST, Embury S, Moohr JW, et al. Cerebrovascular accidents in sickle cell disease: rates and risk factors. Blood. 1998;91:28894.
15. Adams RJ, Brambilla DJ, Granger S, Gallagher D, Vichinsky E, Abboud MR, et al.; STOP Study. Stroke and conversion to high risk in children screened with transcranial Doppler ultrasound during the STOP study. Blood. 2004;103:368994.
16. Wang WC. The pathophysiology, prevention, and treatment of stroke in sickle cell disease. Curr Opin Hematol. 2007;14:1917.
17. National Heart, Lung, and Blood Institute, National Institutes of Health. Clinical alert from the National Heart, Lung, and Blood Institute [press release]. September 18, 1997. Available at: http://www.nhlbi.nih.gov/new/press/nhlb-18a.htm Accessed June 15, 2008
18. Kwiatkowski JL, Granger S, Brambilla DJ, Brown RC, Miller ST, Adams RJ; Stop Trial Investigators. Elevated blood flow velocity in The anterior cerebral artery and stroke risk in sickle cell disease: Extended analysis from the STOP trial. Br J Haematol. 2006;134:3339.
19. Driscoll MC, Hurlet A, Styles L, McKie V, Files B, Olivieri N, et al.. Stroke risk in Siblings with sickle cell anemia. Blood. 2003;101:24014.
20. Hoppe C, Klitz W, Cheng S, Apple R, Steiner L, Robles L, et al.; CSSCD Investigators. Gene interactions and stroke risk in children with sickle cell anemia. Blood. 2004;103:23916.
21. Sebastiani P, Ramoni MF, Nolan V, Baldwin CT, Steinberg MH.Genetic dissection and prognostic modeling of overt stroke in sickle Cell anemia. Nat Genet. 2005;37:43540.
22. Trompeter S, Roberts I. Haemoglobin F modulation in childhood sickle cell disease. Br J Haematol. 2009 Feb;144(3):308-16.
23. Pegelow CH, Adams RJ, McKie V, Abboud M, Berman B, Miller ST, et al. Risk of recurrent stroke in patients with sickle cell disease treated with erythrocyte transfusions. J Pediatr. 1995;126:8969.
24. Dobson SR, Holden KR, Nietert PJ, Cure JK, Laver JH, Disco D, et al. Moyamoya syndrome in childhood sickle cell disease: a predictive factor for recurrent cerebrovascular events. Blood. 2002;99:314450.
25. Moser FG, Miller ST, Bello JA, Pegelow CH, Zimmerman RA, Wang WC, et al. The spectrum of brain MR abnormalities in sickle-cell disease: a report from the Cooperative Study of Sickle Cell Disease. AJNR Am J Neuroradiol. 1996;17:96572.
26. Armstrong FD, Thompson RJ Jr, Wang W, Zimmerman R, Pegelow CH, Miller S, et al. Cognitive functioning and brain magnetic resonance imaging in children with sickle cell disease. Neuropsychology Committee of the Cooperative Study of Sickle Cell Disease. Pediatrics. 1996;97:86470.
27. Miller ST, Macklin EA, Pegelow CH, Kinney TR, Sleeper LA, Bello JA, et al.; Cooperative Study of Sickle Cell Disease. Silent infarction as a risk factor for overt stroke in children With sickle cell anemia: a report from the Cooperative Study of Sickle Cell Disease. J Pediatr. 2001;139:38590.
28. Prengler M, Pavlakis SG, Prohovnik I, Adams RJ. Sickle cell disease: the neurological complications. Ann Neurol. 2002;51:54352.
29. Strouse JJ, Hulbert ML, DeBaun MR, Jordan LC, Casella JF. Primary hemorrhagic stroke in children with sickle cell disease is associated with recent transfusion and use of corticosteroids. Pediatrics. 2006;118:191624.
30. Fullerton HJ, Wu YW, Zhao S, Johnston SC. Risk of stroke in children: ethnic and gender disparities. Neurology. 2003;61:18994.
31. Rothman SM, Fulling KH, Nelson JS. Sickle cell anemia and central nervous system infarction: a neuropathological study. Ann Neurol. 1986;20:68490.
32. Roach ES, Golomb MR, Adams R, Biller J, Daniels S, deVebrer G, et al. Management of Stroke in Infants and Children. A Scientific Statement From a Special Writing Group of American Heart Association Stroke Council and the Council on Cardiovascular Disease in the Young. Stroke. 2008;39:2644-91.
33. Smith ER, McClain CD, Heeney M, Scott RM. Pial synangiosis in patients with moyamoya syndrome and sickle cell anemia: perioperative management and surgical outcome. Neurosurg Focus. 2009 Apr;26(4):E10.
34. Charache S, Terrin ML, Moore RD, Dover GJ, Barton FB, Eckert SV, et al. Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia. Investigators of the Multicenter Study of Hydroxyurea in Sickle Cell Anemia. N Engl J Med. 1995;332: 131722.
35. Colombatti R, Meneghetti G, Emani M, Pierobon M, Sainati L. Primary stroke prevention for sickle cell disease in north-east Italy: the role of ethnic issues in establishing a Transcranial Doppler screening program. Riv Ital Pediatr. 2009 Jun 22;35(1):15.
36. Roberts L, O'Driscoll S, Dick MC, Height SE, Deane C, Goss DE, et al. Stroke prevention in the young child with sickle cell anaemia. Ann Hematol. 2009;88:943-6. .
37. Gulbis B, Haberman D, Dufour D, Christophe C, Vermylen C, Kagambega F, et al. Hydroxyurea for sickle cell disease in children and for prevention of cerebrovascular events: the Belgian experience. Blood. 2005;105:268590.
38. Ware RE, Zimmerman SA, Sylvestre PB, Mortier NA, Davis JS, Treem WR, Schultz WH. Prevention of secondary stroke and resolution of transfusional iron overload in children with sickle cell anemia using hydroxyurea and phlebotomy. J Pediatr. 2004;145:34652.
Recibido: 30 de julio de 2009.
Aprobado: 16 de septiembre de 2009.
José Vargas Díaz. Servicio de Neuropediatría, Instituto de Neurología y Neurocirugía. Calle 29 y D, El Vedado. La Habana, Cuba.
Correo electrónico: jvargas@infomed.sld.cu

