










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Pediatría
versión On-line ISSN 1561-3119
Rev Cubana Pediatr v.82 n.2 Ciudad de la Habana abr.-jun. 2010
Síndrome hemofagocítico secundario en el recién nacido
Secondary hemophagocytosis syndrome in the newborn
Eduardo Morales Mesa,I María de los Ángeles Cubero Rego,II Oneida Reyes Romero,III Regla Broche Cardó,IV María Antonia Pérez Moré,V Nilvia Esther González GarcíaVI
IEspecialista de II Grado en Neonatología. Hospital Pediátrico Universitario «William Soler». La Habana, Cuba.
IIEspecialista de I Grado en Neonatología y Medicina General Integral. Hospital Pediátrico Universitario «William Soler». La Habana, Cuba.
IIIEspecialista de II Grado en Neonatología. Hospital Pediátrico Universitario «William Soler». La Habana, Cuba.
IVEspecialista de I Grado en Neonatología y Medicina General Integral. Hospital Pediátrico Universitario «William Soler». La Habana, Cuba.
VEspecialista de I Grado en Neonatología. Hospital Pediátrico Universitario «William Soler». La Habana, Cuba.
VIEspecialista de I Grado en Neonatología. Hospital Pediátrico Universitario «William Soler». La Habana, Cuba.
RESUMEN
El síndrome hemofagocítico secundario o reactivo es una entidad poco frecuente, de etiología multifactorial, que clínicamente se manifiesta como un cuadro grave y de alta letalidad. Se caracteriza por activación benigna de los macrófagos, asociada a infecciones virales, bacterianas, fúngicas o parasitarias, a inmunodeficiencias y a neoplasias. Se presenta el caso clínico de un neonato varón, de 17 días de vida, que presentó fiebre, manifestaciones catarrales y digestivas. Evolucionó clínicamente con ictericia, hepatoesplenomegalia, trombocitopenia y anemia. Se diagnosticó un citomegalovirus en la orina. Egresó vivo a los 68 días de vida, con regresión del cuadro clínico.
Palabras clave: Recién nacido, citomegalovirus, síndrome hemofagocítico.
ABSTRACT
The secondary or reactive hemophagocytosis syndrome is an uncommon entity clinically manifested by a severe clinical picture and a high mortality rate. It is characterized by a macrophages benign activation, associated with viral, bacterial, fungoid or parasitic infections and immunodeficiencies and neoplams. This is the clinical case of a 17 days male neonate with fever, suffering from cold and digestive manifestations who clinically evolving with jaundice, hepatosplenomegaly and anemia. A cytomegalovirus was diagnosed in urine. Was discharged being alive at 68 days with a regression of clinical picture.
Key words: Newborn, cytomegalovirus, hemophagocytosis sindrome.
INTRODUCCIÓN
La hemofagocitocis se define como la proliferación de las células histiocíticas que conservan su morfología normal, con intensa actividad para fagocitar células hematopoyéticas (leucocitos, eritrocitos, y plaquetas).1
El síndrome hemofagocítico inicialmente fue considerado como un proceso maligno primario llamado histiocitosis maligna o reticulosis medular histiocítica. En 1979, Risdall y cols.1 describieron este síndrome asociado a infección viral y tiempo después otros autores propusieron su asociación a infecciones bacterianas, fúngicas o parasitarias.2,3 Más tarde este síndrome se describió en relación con colagenosis y neoplasias malignas denominándose, síndrome hemofagocítico reactivo (SHR).4
Se presenta el caso clínico con el objetivo de divulgar esta rara enfermedad en el período neonatal, pues en este medio las sepsis neonatales son frecuentes y en múltiples ocasiones presentan un desenlace fatal con signos de «coagulación intravascular diseminada». El conocimiento de los criterios presentes en esta entidad permitiría el pensamiento diagnóstico y la terapéutica precoz y efectiva para los pacientes, con una mayor supervivencia.
PRESENTACIÓN DEL CASO
Recién nacido del sexo masculino, de 17 días de vida, hijo de madre de 19 años con antecedentes patológicos personales de sepsis vaginal tratada en el tercer trimestre. Nació a las 38,5 semanas de edad gestacional, con peso de 2 760 g y Apgar 9/9, y fue egresado de su maternidad a los 3 días. Permaneció asintomático hasta los 16 días de vida, cuando comienza con manifestaciones catarrales y temperatura de 38,5 oC. Al examen físico se le detectó obstrucción nasal moderada con secreción amarilla, tiraje subcostal, murmullo vesicular rudo, abundantes ruidos transmitidos, no estertores, polipnea irregular con frecuencia promedio 68´ y en la otoscopia membranas timpánicas congestivas. Los complementarios del ingreso oscilaron dentro de límites normales e incluyeron radiografía de tórax. Por el examen otoscópico y el cuadro clínico se inició tratamiento antibiótico con penicilina cristalina y amikacina según protocolos.
A las 48 h presentó empeoramiento clínico, gasométrico (pH: 7,26 pCO2 71,2 pO2 66 EB 3,8 SB 27 sat O2 88) y radiológico del cuadro respiratorio y se interpretó como una bronconeumonía (figura 1), por lo que requirió ventilación mecánica. Se sustituyó la penicilina por ceftriaxona y se comenzó con inmunoglobulina en dosis inmunomoduladoras; a las 48 h de ventilación inicia con gran distensión abdominal, contenido gástrico lácteo y deposiciones diarreicas.

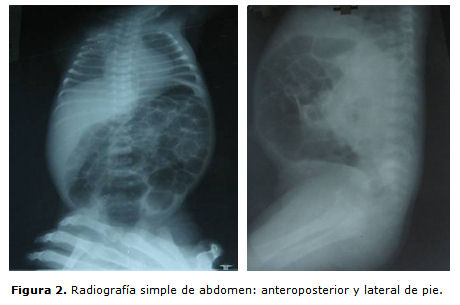
Se realizó radiografía de abdomen de pie anteroposterior y lateral (figura 2), donde se observaron asas intestinales muy dilatadas, con edema. Se interpretó como una enteritis secundaria a la sepsis respiratoria, por lo que se suspendió la vía oral y se decidió cambiar de antibióticos para meronen (11 días) y vancomicina (11 días). A las 72 h de estar sin contenido y desaparecer la distensión abdominal se reinició la vía oral con leche materna, la cual toleró durante 3 días. Reaparece gran distensión abdominal, contenido gástrico sanguinolento y anemia. Se vuelve a suspender la vía oral, se transfunde y se inicia nutrición parenteral que se mantuvo por 5 días; al quinto día se constata íctero intenso, palidez marcada, hepatomegalia de 7 cm y esplenomegalia de 5 cm; mantuvo la distensión abdominal y se halló anemia y petequias generalizadas. Se le realizaron complementarios: hematócrito 0,28, coagulograma: tiempo de protrombina control 12 s, paciente 12 s, kaolín 45 s, plaquetas 1 000 x 109/L, leucograma 10,0x 109 /L , polimorfonucleares 063, linfocitos 047, transaminasa glutámico pirúvica (TGP) 56 UI, transaminasa glutámico oxalacítica (TGO) 60 UI, bilirrubina total elevada a expensas de la directa. En el ultrasonido abdominal se observó pequeña cantidad de líquido libre en cavidad, hígado de 88 mm y bazo que sobrepasaba el polo inferior de riñón izquierdo con una medida 66 mm. Se encuentran células levaduriformes en la orina, por lo que se inició tratamiento con anfotericin B liposomal.
En el hemocultivo crecen una Candida krussein y un citrobacter y se realiza nuevo cambio antibiótico para teicoplamina y tazocin.
Se interconsultó con los hematólogos, por la posibilidad de un síndrome hemofagocítico. En el medulograma se observa la presencia de histiocitos con predominio de hemofagocitos, lo que corroboró la entidad sospechada.
Se comienza el tratamiento, según protocolo, con etoposide, dexametasona, ciclosporina e intacglobin a dosis inmunosupresoras. Además, para completar el estudio se realizaron triglicéridos que se encontraban elevados (1,96mmol/L) al igual que el colesterol 11,9 mmol/L; TGP 81,4UI; urea 9,8 µmol/L; albúmina 31,9 g/L; proteínas totales 73,3 g/L; ferritina > 2 000 ng/mL y fibrinógeno normal. Al mismo tiempo se pensó en la posibilidad de una enfermedad por inclusión citomegálica, por lo que se recogieron muestras de orina y sangre, con resultados positivos de la reacción en cadena de la polimerasa (PCR) en orina para citomegalovirus.
A la semana de tratamiento inmunosupresor se inició tratamiento con ganciclovir parenteral; ya en esos momentos el paciente no presentaba ictericia, el hígado había disminuido de tamaño y la esplenomegalia era de 2 cm, plaquetas en 120 x109/L, conteo de neutrófilos dentro de límites normales y hemoglobina de 100 g/L; a los 4 días del tratamiento con ganciclovir, las plaquetas vuelven a caer a 20x 109/L, el conteo de neutrófilos a 118 y el hematócrito a 0,22. Por esta razón, se suspendió el tratamiento durante 4 días y se reinició nuevamente, pero fue interrumpido por segunda ocasión a los 4 días ya que el paciente presentó trombocitopenia y neutropenia.
A las 72 h de tratamiento inmunosupresor según protocolo establecido para la enfermedad comenzó con hipertensión arterial, por lo que se realizó ecocardiograma, donde no se observaron alteraciones y se inició tratamiento con captopril y furosemida; al mantener cifras de tensión arterial media entre 80 y 100 mm Hg se le añadió propanolol y se logró el control de la tensión arterial. Egresa vivo a las 4 semanas de tratamiento inmunosupresor, con un peso de 3 250g, una hemoglobina de 100 g/L, conteo de plaquetas en 250 x 109/L, y leucograma en 11,0 x 109/L leucocitos, con 0,63 polimorfonucleares, 0,33 linfocitos y 0,01 monocito. Con lactancia materna y seguimiento por hematología para concluir tratamiento inmunosupresor según protocolo.
DISCUSIÓN
Basado en los criterios diagnósticos establecidos por la Sociedad de Histiocitosis en 1991; en este paciente se planteó un síndrome hemofagocítico reactivo o secundario (SHR), a una sepsis, ya que no se encontraron antecedentes familiares de consanguinidad o de enfermedad familiar similar.6
El síndrome hemofagocítico no pensado, ni diagnosticado, tiene importantes implicaciones pronósticas para el paciente.
Criterios para su diagnóstico: 6
- Clínicos:
- Fiebre (duración mayor o igual a 7 días con picos > 38,5 ºC).
- Esplenomegalia (mayor o igual a 3 cm por debajo del borde costal).
- Laboratorio:
- Citopenia (que afecte dos o más de tres líneas celulares en sangre periférica, que no sea causada por médula ósea hipocelular o displásica).
- Anemia (hemoglobina < 900 g/dL).
- Trombocitopenia (recuento plaquetario < 100 x 109).
- Neutropenia (< 1,0 x 109 g/L).
- Hipertrigliceridemia-hipofibrinogenemia (triglicéridos en ayuno mayor o igual a 2,0 mmol/L o más de 3 veces del valor normal para la edad, fibrinógeno menor igual a 1,5 g/L o menor o igual a 3 veces el valor normal).
- Histopatológicos:
- Hemofagocitosis en la médula ósea, el bazo o los nódulos linfáticos, sin datos de malignidad.
Aunque el síndrome hemofagocítico secundario no es una patología frecuente en pediatría, pareció importante la divulgación de este paciente con la finalidad de sensibilizar a los neonatólogos sobre la sospecha clínica precoz de la entidad.
La aparición del síndrome, asociado a una condición subyacente, por lo general, infecciosa, complica gravemente la evolución y el manejo del paciente, con una alta mortalidad, lo que podría evitarse con el inicio de un tratamiento adecuado y oportuno.
En Cuba se conocen, mediante comunicación verbal (Villegas D. 2009) dos casos diagnosticados con esta entidad, en el período neonatal, ambos fallecidos a pesar del tratamiento inmunosupresor impuesto según protocolo.
En el estudio de Morales Ferrer la edad de mayor presentación fue en menores de 3 años, sin predominio de sexo; el 87,5 % (21 casos) fallecieron sin diagnóstico clínico de síndrome hemofagocítico y solo el 12,5 % fueron diagnosticados en vida.1
Janka y colaboradores, en un estudio de 219 niños, observaron que la mortalidad de enfermos con síndrome hemofagocítico fue mayor del 50 % en pacientes hasta los 3 años de edad, del 38 % en mayores de 3 años y el pronóstico fue especialmente malo en menores de 1 año.7
La enfermedad se produce por la proliferación exagerada y activación histiocítica que normalmente se asocia a infección viral sistémica por ejemplo a herpes virus, virus de la varicela zoster, herpes zoster, citomegalovirus, virus de Epstein-Barr, adenovirus, parvovirus, hepatitis B, sarampión e incluso VIH. En ocasiones a infecciones bacterianas, fúngicas, protozoarias, trastornos autoinmunitarios y enfermedades malignas.6, 8,9
En nuestro paciente se aisló el citomegalovirus en orina, una Candida krussein y un citrobacter.
El manejo debe incluir una sospecha precoz de esta condición clínica y el tratamiento adecuado de la causa subyacente, lo cual influye en la evolución. La mortalidad varía del 20 al 40 %, cuando se asocia a infección y aumenta a casi un 100 % cuando se asocia a otras causas, especialmente patologías malignas.9,10
Nos quedará siempre la duda si la aparición del síndrome fue secundario a una sepsis bacteriana o micótica, o si el citomegalovirus desencadenó el desarrollo de la enfermedad, ya que el paciente ingresó con una infección respiratoria alta, tuvo una estadía hospitalaria de 15 días antes de realizarle el diagnóstico y recibió nutrición parenteral; además de ser politransfundido con diversos hemoderivados.
Lo que si debe tenerse presente es que las infecciones virales del tipo de los citomegalovirus, pueden debutar desde el nacimiento o en la segunda o tercera semana de vida con manifestaciones respiratorias aparentemente banales.
El diagnóstico y tratamiento precoz del síndrome hemofagocítico secundario o reactivo, puede cambiar el pronóstico oscuro de estos pacientes para lograr la supervivencia.
REFERENCIAS BIBLIOGRÁFICAS
1. Morales FG, Durán PM A, Córdova RS. Síndrome hemofagocítico reactivo. Rev Med Hosp Gen Mex. 2002;65(4):207-12.
2. Aouad A, Dan EM, Alangade JG. Diagnosis: reactive hemophagocytic syndrome caused by CMV infection. Clin Infect Dis. 1998;26:1295.
3. Wong FK, Chan CKJ, Ha YS, Wong WH. Reactive hemophagocytic syndrome in childhood- frequent occurrence of atypical mononuclear cells. Hematol Oncol. 1994;12:67-74.
4. Herrero HA, Ramírez JS, García ME, Martínez VA. Síndromes hemofagocíticos. An Esp Pediatr. 1998;49:230-6.
5. Mourad Tiab F, Mechinaud, Harousseau JL. Haemophagocytic syndrome associated with infections. Baillière's Clinical Haematogy. 2000;13:163-78.
6. Henter JI, Elinder G, Ôst A, FHL Study Group of the Histiocyte Society. Diagnostic guidelines for hemophagocytic lymphohistiocytosis. Semin Oncol. 1991;1829:33.
7. Janka G, Imashuku S, Elinder G, Schneider M, Henter J. Infection and malignancy associated hemophagocytic syndromes. Hematol Oncol Clin North Am. 1998;12:67-74.
8. Filipovich AH. Hemophagocytic lymphohistiocytosis. Immunol Allergy Clin North Am. 2002;22:2.
9. Verdugo P, Rodríguez N, Tordecilla J, Soto V. Síndrome hemofagocítico secundario en Pediatría. Experiencia clínica en ocho casos. Rev Chil Pediatr. 2005;76(4):397-403.
10.Strauss R, Neureiter D: Multifactorial risk analysis of bone marrow histiocytic hyperplasia with hemophagocytosis in critically ill medical patients. A post-mortem nclinicopathologic analysis. Crit Care Med. 2004;32:1316-21.
Recibido: 17 de febrero de 2010.
Aprobado: 26 de marzo de 2010.
Eduardo Morales Mesa. Hospital Pediátrico Universitario «William Soler». San Francisco 10112, Altahabana. La Habana, Cuba.
Correo electrónico: eduarm@infomed.sld.cu

