










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Pediatría
versión impresa ISSN 0034-7531versión On-line ISSN 1561-3119
Rev Cubana Pediatr v.83 n.2 Ciudad de la Habana abr.-jun. 2011
Síndrome del QT largo
Syndrome of long Q-T interval
Michel Cabrera Ortega,I Francisco Javier Ozores Suárez II
IMáster en Urgencias Médicas. Especialista de I Grado en Cardiología. Especialista de I Grado en Medicina General Integral. Instructor de Pediatría. Cardiocentro Pediátrico «William Soler». La Habana, Cuba.
IIEspecialista de I y II Grado en Cardiología. Asistente de Pediatría. Cardiocentro Pediátrico «William Soler». La Habana, Cuba.
RESUMEN
El síndrome del QT largo congénito de tipo Romano-Ward es una canalopatía arritmogénica poco frecuente, caracterizada por una grave alteración en la repolarización ventricular y traducida en el electrocardiograma por un alargamiento del intervalo QTc. Se documenta el caso de una paciente con síndrome del QT largo congénito de tipo 1, asintomática, con antecedentes familiares de muerte súbita y síndrome del QT largo, a quien se le realizó estudio ecocardiográfico, prueba ergométrica, detección de potenciales tardíos y dispersión del QT como complementos diagnósticos y estratificadores de riesgo. Se prescribió tratamiento farmacológico y semanas después se valoró su efectividad.
Palabras clave: Síndrome del QT largo, canalopatía arritmogénica, prueba de esfuerzo, bloqueador de los receptores β.
ABSTRACT
The syndrome of congenital long Q-T interval (Q-T-i) of Romano-Ward type is an uncommon arrthymic channel disease, characterized by a severe alteration in ventricular repolarization and translated in the electrocardiogram by a lengthening of QTc interval.
This is the case of a female patient presenting with type 1 congenital long Q-T, asymptomatic, with family history of sudden death and syndrome of long Q-T; an echocardiography study, ergometer test, late potentials detection and Q-T dispersion as diagnostic complements and risk stratification. Drug therapy was prescribed and several weeks later its effectiveness was assessed.
Key words: Long Q-T syndrome, arrhythmic channel disease, endurance test, β-receptor blockers.
INTRODUCCIÓN
El síndrome de QT largo (SQTL) congénito de variante Romano-Ward es una canalopatía arritmogénica poco frecuente (1/5 000-10 000), caracterizada por una grave alteración en la repolarización ventricular traducida en el electrocardiograma (ECG) por un alargamiento en el intervalo QT, que predispone a arritmias ventriculares malignas y muerte súbita.
Se han descrito 12 subtipos y tiene sus bases en la expresión de mutaciones en los genes responsables del correcto funcionamiento de los canales iónicos que generan el potencial de acción transmembrana.1
Se presenta un caso con SQTL de tipo 1 y se realiza una actualización breve del tema.
PRESENTACIÓN DEL CASO
Paciente del sexo femenino, de 5 años, con antecedentes de salud, que es ingresada para estudio por antecedentes familiares de muerte súbita, relacionada con SQTL. Al examen físico no se detectó alteración alguna.
Se le realizó ECG de 12 derivaciones y se detectó un intervalo QT corregido (QTc) por la fórmula de Bazzet, de 465 ms, 481 ms y 465 ms, en tres latidos consecutivos, con un promedio de 475 ms (figura 1).
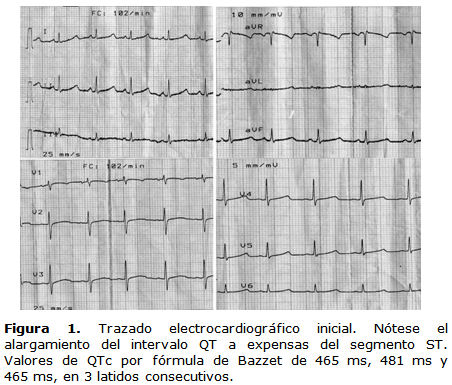
Se indicó un ecocardiograma que reveló estructuras cardíacas normales. Se realizó una prueba ergométrica y se evidenció una frecuencia cardíaca máxima de 160 lpm y un alargamiento del QTc en esfuerzo máximo y recuperación temprana de 516 ms. Como complementos de la estratificación de riesgo se valoró la presencia de potenciales tardíos y dispersión del QT, y ambos estudios fueron positivos. Se aplicó la puntuación de Schwartz1 para el diagnóstico de síndrome de QT largo, con una probabilidad alta al obtener 4,5 puntos. Se concluyó entonces que se trataba de un síndrome de Romano-Ward con expresión fenotípica de un SQTL1.
Se prescribió tratamiento farmacológico con propanolol, que se inició con dosis de 1 mg/(kg·d) y se incrementó paulatinamente hasta 2,5 mg/(kg·d). Como medida complementaria se recomendó evitar la práctica de deportes competitivos, natación o ejercicio vigoroso. Tras tres semanas de iniciado el tratamiento se repitió la prueba ergométrica y se evidenció un frecuencia cardíaca máxima de 130 lpm y un incremento del QTc al esfuerzo máximo y recuperación temprana de 470 ms. El ECG mostró una normalización del intervalo QTc con valores de 447 ms, 417 ms y 436 ms en tres latidos consecutivos con un promedio de 433 ms (figura 2).

DISCUSIÓN
Existen dos variedades del SQTL congénito: el síndrome de Jervell-Lange Nielsen, que se acompaña de sordera neuronal congénita y alto riesgo de muerte súbita, y el síndrome de Romano-Ward, en el que los pacientes no presentan trastornos auditivos y la gravedad de la enfermedad es variable.1,2 Dentro de este último se han descrito 12 formas de acuerdo con los 12 genes asociados a la enfermedad. Sin embargo, el SQTL1, SQTL2 y SQTL3 explican el 65 % de los casos y se diferencian entre sí por la expresión fenotípica.2
El diagnóstico de esta paciente se concluyó como un SQTL1, pero el ECG no era típico de este subtipo ya que mostraba un alargamiento del QTc a expensas más del segmento ST que de la onda T. A pesar de que el trazado electrocardiográfico tiene una sensibilidad de hasta de un 85 %2 para el SQTL1, nos basamos en otros elementos para no incluir a la paciente como un SQTL3, a saber: los antecedentes revelan el advenimiento de la muerte súbita en tres familiares durante la actividad física, lo cual caracteriza al SQTL1 en el 62 % de los casos. Se debe destacar que la respuesta durante la prueba ergométrica no es típica de un SQTL3, debido a que en este subtipo al esfuerzo máximo el intervalo QTc se acorta o no sufre variaciones como respuesta fisiológica al ejercicio, mientras que los pacientes con un SQTL1, como esta paciente, además de no llegar a la frecuencia cardíaca máxima calculada para la edad alargan el intervalo QTc (sensibilidad del 77 %, especificidad del 90 %).3
La positividad de marcadores eléctricos no invasivos (potenciales tardíos, dispersión del QT) como indicadores de heterogeneidad de la repolarización miocárdica y la probabilidad de que se desencadenen arritmias ventriculares malignas,4 junto al antecedente familiar de muerte súbita y la edad al diagnóstico, son elementos para tener en cuenta en un paciente asintomático, como en este caso, en el momento de decidir el inicio del tratamiento con un bloqueador de los receptores β, de acuerdo a las guías consultadas.5,6 A ello se suman las modificaciones en el estilo de vida.
La favorable respuesta de nuestra paciente tras 3 semanas de tratamiento antiarrítmico fue un indicador más de que se trataba de un SQTL1. La respuesta del SQTL1 a los bloqueadores de los receptores β ha logrado que cerca del 90 % de los pacientes estén libres de síntomas (síncope, muerte súbita) en un seguimiento de 5,4 años2,5,6 y que un número reducido de casos requieran la implantación de un cardiodesfibrilador automático implantable, un marcapaso o la denervación simpática cardíaca izquierda como terapias alternativas.2
REFERENCIAS BIBLIOGRÁFICAS
1. Gómez-Gómez M, Danglot-Banck C, Santamaría-Díaz H. Síndrome de QT largo en pediatría. Rev Mex Pediatr. 2008;75(3):121-31.
2. Ruan Y, Liu N, Napolitano C and Priori S. Therapeutic strategies for long-qt syndrome. Does the molecular substrate matter? Circulation: Arrhythmia and Electrophysiology. 2008;1:290-7.
3. Walker BD, Krahn AD, Klein GJ, Skanes AC, Yee R. Burst bicycle exercise facilitates diagnosis of latent long QT syndrome. Am Heart J. 2005;150:1059-63.
4. Goldenberg I, Moss AJ, Peterson DR, McNitt S, Zareba W, Andrews ML et al. Risk factors for aborted cardiac arrest and sudden cardiac death in children with congenital long-QT syndrome. Circulation. 2008;117:2184-91.
5. Lehnart SE, Ackerman MJ, Benson DW, Brugada R, Clancy CE, et al. Inherited Arrhythmias: A National Heart, Lung, and Blood Institute and Office of Rare Diseases Workshop Consensus Report about the diagnosis, phenotyping, molecular mechanisms, and therapeutic approaches for primary cardiomyopathies of gene Mutations affecting ion channel function. Circulation. 2007;116;2325-45.
6. Zipes DP, Camm AJ, Borggrefe M, Buxton AE, Chaitman B, Fromer M, Gregoratos G, et al. ACC/AHA/ESC 2006 Guidelines for management of patients with Ventricular Arrhythmias and the prevention of Sudden Cardiac Death. A Report of the American College of Cardiology/American Heart Association task force and the European Society of Cardiology Committee for practice guidelines. J Am Coll Cardiol. 2006;48:247-346.
Recibido: 3 de marzo de 2011.
Aprobado: 16 de marzo de 2011.
Michel Cabrera Ortega. Cardiocentro Pediátrico «William Soler». Calle 100 y Perla. Altahabana, Boyeros. La Habana, Cuba. CP 10800.
Correo electrónico: michel@cardiows.sld.cu

