










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Pediatría
versión impresa ISSN 0034-7531
Rev Cubana Pediatr vol.83 no.3 Ciudad de la Habana jul.-set. 2011
PRESENTACIÓN DE CASOS
Encefalomenigocele atrésico parietal
Parietal atresic encephalomeningocele
Liliana Rivera Oliva,I Yanett Sarmiento Portal,II Odalys Hernández León,III María Elena Portal Miranda,IV Yunitseis Martínez VergaraV
IEspecialista de I Grado en Neonatología. Máster en Atención Integral al Niño. Asistente. Hospital Universitario "Abel Santamaría Cuadrado". Pinar del Río, Cuba.
IIEspecialista de I Grado en Medicina General Integral y Neonatología. Máster en Atención Integral al Niño. Asistente. Hospital Universitario "Abel Santamaría Cuadrado". Pinar del Río, Cuba.
IIIEspecialista de I Grado en Neurocirugía. Hospital Universitario "Abel Santamaría Cuadrado". Pinar del Río, Cuba.
IVEspecialista de II Grado en Neonatología. Máster en Atención Integral al Niño. Asistente. Hospital Universitario "Abel Santamaría Cuadrado". Pinar del Río, Cuba.
VResidente de 2do. año de Neonatología. Hospital Universitario "Abel Santamaría Cuadrado". Pinar del Río, Cuba.
RESUMEN
El encefalocele es una anomalía congénita rara, en la que una porción del encéfalo protruye a través de un orificio craneal (evaginación), generalmente situado en la línea media. Clínicamente se caracteriza por una masa epicraneal, de consistencia blanda, muchas veces acompañada de trastornos psicomotores, convulsiones y trastornos de la visión. Se presenta el caso de un recién nacido con diagnóstico de encefalomeningocele atrésico parietal, intervenido quirúrgicamente y con evolución satisfactoria.
Palabras clave: encefalocele, cefalocele.
ABSTRACT
The encephalocele is a uncommon congenital anomaly where a portion of encephalon protrudes through a cranial orifice (evagination), generally located in the middle line. Clinically, it is characterized by a soft epicranial mass often accompanied or psychomotor disorders, convulsions and vision disorders. This is the case of a newborn diagnosed with parietal atresic encephalomeningocele operated on with a satisfactory evolution.
Key words: encephalocele, cephalocele.
INTRODUCCIÓN
El encefalocele es el defecto del tubo neural menos frecuente. Se presenta entre un caso de cada 2 000 a 6 000 nacidos vivos, pero su incidencia varía considerablemente según los diferentes estudios, aunque es, al parecer, más frecuente en México, en países de origen celta y ciertos países del sureste asiático como Indonesia, Malasia y Tailandia, donde llega a alcanzar una frecuencia de 1 por cada 5 000 nacidos vivos. En España se estima una prevalencia global de 0,80 por 10 000 recién nacidos vivos.1,2
Este defecto puede ir acompañado de alteraciones craneofaciales como: microcefalia, fisura palatina, y alteraciones neurológicas, como la hidrocefalia y la espina bífida. Según el grado o intensidad de la evaginación, existen varias formas clínicas ordenadas de menor a mayor gravedad: cráneo bífido oculto, meningocele craneal, encefalomeningocele, cefaloceles atrésicos (formas involucionadas o abortadas que contienen tejido neural o glial), hidrancefalocele y encefalocistomeningocele. Clínicamente se caracteriza por una masa epicraneal, de consistencia blanda, muchas veces acompañada de trastornos psicomotores, convulsiones y trastornos de la visión.2-4 Ante la presencia de un recién nacido con esta anomalía congénita, surge la motivación a realizar una revisión sobre el tema.
PRESENTACION DEL CASO
Recién nacido, de sexo masculino, raza blanca, hijo de madre de 37 años, sana, G1 P0 A0, serología no reactiva, grupo y Rh O+, edad gestacional de 39,1 semanas, nacido por cesárea debido a sufrimiento fetal agudo, tiempo de rotura de membranas de 6 h, líquido amniótico claro, cordón y placenta normal. Apgar 9-9 puntos y peso al nacer 3 000 g.
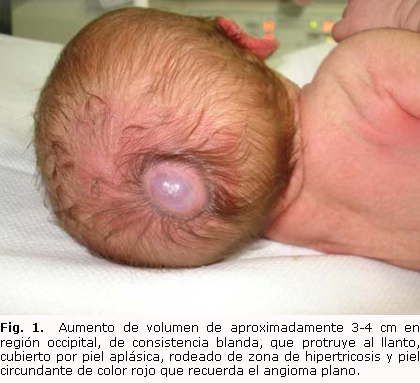
Al examen físico se constata polidactilia axial derecha y a nivel del occipital. Próximo a la fontanela posterior se observa aumento de volumen de aproximadamente 3-4 cm, de consistencia blanda, que protruye al llanto, cubierto por piel aplásica, rodeado de zona de hipertricosis y piel circundante de color rojo, que recuerda el angioma plano (figuras 1 y 2). El resto del examen físico es normal. Estas características descritas sugieren el diagnóstico de un defecto de cierre del tubo neural cerrado.
Se realiza interconsulta con neurocirugía, radiología y genética clínica. Se realiza ultrasonido de cráneo, en el que no se definen alteraciones intracraneales. En el ultrasonido de partes blandas se observa, a nivel de la línea media en región occipital, una imagen relativamente ecolúcida de aproximadamente 4 cm, con ecos finos en su interior, pero no se observan giros ni circunvoluciones. En el TAC de cráneo se observa cavum del septum pelúcido, y no se define relación entre tejido cerebral y la lesión de partes blandas ni otras alteraciones de tipo malformativo.
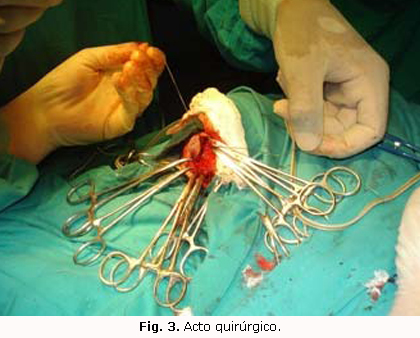
El resto de los complementarios, incluyendo ultrasonido abdominal, Rx tórax, hemograma, glicemia y gasometría, fueron normales. Se decide realizar intervención quirúrgica, con incisión perpendicular a la lesión y se visualiza en el interior la presencia de líquido cefalorraquídeo, duramadre, aracnoides, vaso de aspecto venoso que drena al seno sagital superior y pedículo o tracto fibroso que se introduce intradural a través del orificio meníngeo. Se concluye el diagnóstico de encefalomeningocele atrésico parietal (figura 3), se le opera, y la evolución posoperatoria fue satisfactoria, sin compromiso neurológico, y egresó el paciente con 11 días de vida.
DISCUSIÓN
La causa precisa de estas malformaciones o defectos del tubo neural aún es desconocida. La literatura revisada sugiere etiología multifactorial, aunque hay coincidencia en la mayoría de los trabajos publicados de que el déficit de ácido fólico en la dieta, la ingestión de ácido valproico durante el embarazo, y los antecedentes familiares serían los más influyentes.3-5 Se estima que aproximadamente el 10 % de los defectos del tubo neural son causados por mutaciones genéticas o alteraciones cromosómicas, ya que se ha visto una alta incidencia en hermanos de niños con esta enfermedad.1
Los encefaloceles se localizan en la región occipital en el 75 % de los casos, y en menor proporción, alrededor del 15 %, pueden localizarse en región parietal frontal y sincipital (sincipucio es la parte anterosuperior de la cabeza), y estos últimos se subclasifican por su localización en: nasofrontal, nasoetmoidal y nasoorbital.6
En el caso del encefalomeningocele el contenido típico de la herniación es líquido cefalorraquídeo y tejido neural que se conecta al cerebro a través de un estrecho pedículo; la cubierta del saco herniario puede variar desde una capa bien formada con piel y cabellos, a una delgada capa meníngea, por lo que la lesión puede estar totalmente cubierta por piel, o alternar con zonas desprovistas de esta, que dejan el tejido nervioso al descubierto.1 En el caso que se presenta el defecto se encontraba cubierto por piel aplásica, y se describe el pedículo que lo conecta al tejido cerebral.
El diagnóstico clínico de sospecha se refuerza mediante la maniobra de trans iluminación del saco, que puede poner de manifiesto la presencia de tejido neural. Está indicado realizar una radiografía simple de cráneo y de columna cervical para definir la anatomía de las vértebras, así como una resonancia magnética nuclear para conocer el contenido del saco herniario. La evaluación correcta de un encefalocele debe incluir un examen físico completo para detectar otras enfermedades posiblemente asociadas, así como la práctica de doppler color, que permite apreciar estructuras vasculares en su interior.1,2,4 El diagnóstico diferencial debe tener en cuenta el higroma quístico, en el que no existe ningún defecto óseo, el edema de la calota, teratomas (tumores mixtos complejos, en los que los tejidos múltiples se disponen en órganos diferenciados) y otras anomalías congénitas como anencefalia, hendidura quística braquial, hemangioma y sarcoma mesenquimático.1
El pronóstico es variable, en función, por un lado, del tamaño, la localización y el tipo de tejido cerebral herniado; pero por otro, del número, tipo y severidad de las malformaciones asociadas. Los lactantes con encefalocele tienen más riesgo de presentar una hidrocefalia (acumulación de líquido en el encéfalo) por estenosis (estrechez patológica de un conducto) del acueducto, una malformación de Chiari, o un síndrome de Dandy Walker.1
Aproximadamente la mitad de los pacientes con encefalocele occipital tienen inteligencia normal o levemente disminuida según otros autores. Los encefaloceles parietales siempre están asociados a otras malformaciones cerebrales, y el 40 % de los casos presentan retraso mental. En términos generales la supervivencia varía según las series publicadas, y oscila entre un 60 hasta un 80-90 % en los casos más favorables, siendo mejor cuando el encefalocele es anterior.1,3
La determinación de niveles de alfafetoproteína materna y la realización de ecografía prenatal, permiten el diagnóstico intraútero, que contribuye a un tratamiento más apropiado del paciente y posibilita decantar otras malformaciones y la planificación del tratamiento. La imagen ecográfica del encefalocele consiste en una masa de tejido asociada siempre a un defecto óseo a través del cual se produce la herniación.1 El tratamiento es quirúrgico, y debe ser tratado interdisciplinariamente. La mayoría de los encefaloceles deben corregirse, incluso los más grandes, ya que pueden eliminarse sin provocar incapacidad funcional importante, siendo necesaria la corrección quirúrgica urgente cuando la lesión es abierta, es decir, que no está cubierta por piel.1
La prevención de los defectos en el tubo neural se consigue mediante tratamiento con suplementos orales diarios de ácido fólico, suministrados durante el tiempo que transcurre entre la planificación del embarazo y la semana 12 de gestación, por lo que se aconseja comenzar este tratamiento desde el momento en el que se pretenda un embarazo.4-6 En Chile se reporta una disminución de la incidencia de defectos de cierre del tubo neural tras suplementación con ácido fólico de 1,56 a 0,8-0,9 x 1 000 nacidos vivos.7
REFERENCIAS BIBLIOGRÁFICAS
1. Izquierdo M, Avellaneda A. Encefalomeningocele. Asociación Española para el Registro y Estudio de las Malformaciones Congénitas. (ASEREMAC) [monografía en interntet] 2004. [citado el 22 de noviembre de 2010]. Disponible en: http://iier.isciii.es/er/prg/er_bus2.asp?cod_enf=890
2. Encefalocele [homepage en internet]. [citado el 22 de noviembre de 2010]. Disponible en: http://www.webespecial.com/sindromes/introduccion.htm
3. Gelabert M, García A, Fernández J, Martínez R. Meningocistocele orbitario. Neurocirugía. 2003;14:145-9.
4. Otárola BD, Rostion A. Desarrollo embrionario y defectos de cierre del tubo neural. Rev Ped Elec. 2007;4(3):34-43.
5. Tsunenobu T, Frances M. Folate and human reproduction. Am J Clin Nutr. 2006;83:993-1 016.
6. Finnell RH, Gould A, Spiegelstein O. Pathobiology and genetics of neural tube defects. Epilepsia. 2003;44(3):14-23.
7. Ministerio de Salud. Chile. Defectos de cierre del tubo neural. Guías nacionales de Neonatología [homepage en internet]. 2005. [citado el 26 de noviembre de 2010] Disponible en: http://www.prematuros.cl/guiasneo/defectostuboneural.htm
Recibido: 13 de enero de 2011.
Aprobado: 13 de junio de 2011.
Liliana Rivera Oliva. Hospital Universitario "Abel Santamaría Cuadrado". Gerardo Medina 2 A, reparto Montequín. Pinar del Río, Cuba. Correos electrónicos: ff2004@has.sld.cu


{kind=link}
{kind=link}
{kind=link}