










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Pediatría
versión impresa ISSN 0034-7531
Rev Cubana Pediatr vol.85 no.2 Ciudad de la Habana abr.-jun. 2013
PRESENTACIÓN DE CASO
Agenesia sacra asociada a disrrafismo espinal e hidrocefalia
Sacra agenesia associated to spinal dysraphism and hydrocephaly
Dra. Lisett Hernández León, Dra. Odalys Hernández León, MSc. Dra. Noemí Bárbara Cabrera Domínguez, MSc. Dra. Ivón Aimé Sánchez Monterrey, MSc. Dra. Yanett Sarmiento Portal, MSc. Dra. Angelicia Crespo Campo
Hospital "Abel Santamaría". Pinar del Río, Cuba.
RESUMEN
Introducción: la agenesia sacra es una malformación congénita rara que forma parte del síndrome de regresión caudal. Se caracteriza por un grupo de anomalías en las cuales la columna caudal está ausente. Esta enfermedad es la malformación más frecuente en los hijos de madres diabéticas, además se ha relacionado con otros factores predisponentes, como deficiencias de ácido fólico, de vitaminas, uso de insulina en el embarazo, e incluso, la hipoxia. Entre un 30-40 % de pacientes con agenesia sacra completa, pueden tener asociado un mielomeningocele, y el desplazamiento de las raíces nerviosas empeora los trastornos neurológicos. En estos casos, la hidrocefalia, muchas veces también asociada a malformación Chiari tipo II, está ya presente al nacer.
Caso clínico: se presenta el caso de un neonato con agenesia sacra asociada a disrrafismo espinal e hidrocefalia. La intervención quirúrgica fue precoz, se le colocó derivación ventrículo peritoneal y se realizó la reparación del defecto del tubo neural. La evolución posoperatoria fue favorable, aunque persistieron los déficits neurológicos preoperatorios.
Conclusiones: no se hallaron factores predisponentes en este paciente y el análisis del cariotipo fue normal. Las anomalías óseas de miembros inferiores fueron las más llamativas, así como la presencia de hidrocefalia asociada a malformación Chiari tipo II y mielomeningocele. El tratamiento a estos casos requiere de un enfoque multidisciplinar, y la reparación quirúrgica del mielomeningocele debe ser precoz para conseguir una evolución favorable. Las formas graves pueden ocasionar una muerte temprana neonatal, en cambio, los niños que sobreviven, generalmente presentan inteligencia normal.
Palabras clave: síndrome de regresión caudal, agenesia sacra, displasia caudal, defecto abierto del tubo neural, disrrafia espinal, malformación Chiari tipo II.
ABSTRACT
Introduction: sacra agenesia is a rare congenital malformation as part of the caudal regression syndrome. It is characterized by a group of anomalies in which the caudal cord is absent. This disease is the most common malformation found in children from diabetic mothers but it has also been related to other predisposing factors such as folic acid deficiencies, vitamin deficiencies, use of insulin at pregnancy and even hypoxia. Thirty to forty percent of patients with complete sacra agenesia can also have myelomeningocele, and the displacement of nerve roots worsens the neurological disorders. In these cases, hydrocephaly, many times associated to Chiari malformation type II, is also present at birth.
Clinical case: a neonate with sacra agenesia associated to spinal dysraphism and hydrocephaly. Surgical intervention was performed early, a peritoneal ventricular derivation was placed and the neural tube defect was repaired. The post-surgery evolution was favorable, but the preoperative neurological deficits persisted.
Conclusions: there were no predisposing factors in this patient and the analysis of the cariotype was normal. The bone anomalies of the lower members were the most remarkable aspects as well as the hydrocephaly associated to Chiari malformation type II and myelomeningocele. The treatment of these cases requires multidisciplinary approach and surgical repair of the myelomeningocele at early phase to achieve favorable evolution. The most severe forms can cause early neonatal death; however, those surviving children generally present normal intelligence coefficient.
Key words: caudal regression syndrome, sacra agenesia, caudal dysplasia, open neural tube defect, spinal dysraphia, Chiari malformation type II.
INTRODUCCIÓN
La agenesia lumbosacra, también denominada agenesia sacra o agenesia caudal, es una malformación congénita rara que forma parte del síndrome de regresión caudal.1-3 Se caracteriza por un grupo de anomalías, en las cuales la columna caudal está ausente, y varían desde la agenesia parcial aislada de la columna sacrococcígea, a deformidades más graves que incluyen la región lumbar del raquis.4
Duhamel, en 1961, propone el término síndrome de regresión caudal (SRC) para agrupar un espectro de malformaciones congénitas en las que concomitan las alteraciones esqueléticas (agenesia lumbosacra), combinadas con deformidades variables de los miembros inferiores y malformaciones del tracto digestivo, genitourinario, así como déficit neurológico.1 El espectro de este síndrome va desde la asintomática aplasia coccígena, hasta la ausencia de vértebras sacra, lumbar y torácicas, con severos defectos esqueléticos y neurológicos asociados. Como la mayoría de las anomalías incluyen solo el sacro, el término de agenesia sacra ha sido usado como sinónimo de agenesia caudal o regresión caudal.4,5
La agenesia sacra completa puede tener severas deformidades y asociarse a defectos de cierre del tubo neural y malformaciones de la médula espinal, como son, la estenosis del saco dural, el estrechamiento del canal óseo, la diastematomielia, el mielomeningocele, la médula anclada y las bandas aracnoideas adhesivas.5 Entre un 30-40 % de pacientes con agenesia caudal, pueden tener asociado un mielomeningocele, y el desplazamiento de las raíces nerviosas empeora los trastornos neurológicos; además, la tumoración quística suele estar cubierta por una delgada membrana meníngea que se desgarra con facilidad, lo que conlleva a un elevado riesgo de infección que se presenta entre un 10-15 % de los casos.6,7 La asociación de la agenesia sacra y el mielomeningocele puede no ser mortal, pero cuando es grave provoca alteraciones motoras severas. En estos casos la hidrocefalia, también asociada a una malformación Chiari tipo II, está ya presente al nacer.6,7
A continuación, se presenta el caso de un neonato con agenesia sacra unilateral, asociada a un defecto abierto del tubo neural e hidrocefalia concomitante, que fue tratado en nuestro Servicio, y se incluye una discusión de este a partir de una revisión de la literatura científica.
PRESENTACIÓN DEL CASO
Recién nacido del sexo masculino, hijo de madre multípara, que nace por cesárea debido a presentación pelviana a las 39,4 semanas, con un peso de 4 100 g y Apgar 8:9. El examen físico revela:
- Implantación baja de las orejas con tercio inferior facial amplio y aplanado.
- Tórax pequeño.
- Perímetro cefálico de 51 cm, fontanela anterior abombada, tensa y asociada a diastasis de las suturas.
- Actitud en semiflexión de las extremidades con pie varo equino bilateral severo.
- Regiones glúteas achatadas con acortamiento del surco interglúteo.
- Postura cifoescoliótica de la columna vertebral dorsal y defecto del tubo neural a nivel dorsolumbar, de aproximadamente 7 cm en sentido cefalocaudal, con exposición de la placa cubierta por membranas de apariencia aracnoidal (transparentes) sin salida de líquido cefalorraquídeo (LCR), rodeada circunferencialmente por piel displásica (figura 1).
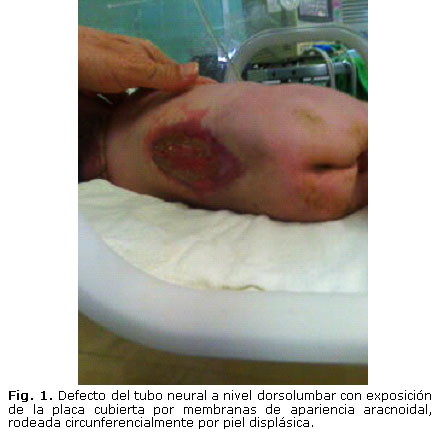
El examen neurológico evidencia paraplejía fláccida bilateral, arreflexia de miembros inferiores sin control esfinteriano, y evidencias de anestesia en ambos miembros inferiores. Inicialmente se cubre el defecto neural con esponja hemostática y se irriga periódicamente con solución ringer lactato. En ultrasonografía transcraneal (USG) se identifica gran dilatación simétrica de todo el sistema ventricular, y en rayos x de columna vertebral agenesia sacra.
Se realiza resonancia magnética nuclear (RMN) craneal y de todo el eje espinal. En la figura 2 se observa que el paciente presenta una hidrocefalia triventricular extrema asociada a un IV ventrículo con dimensiones normales. La fosa posterior presenta marcada reducción volumétrica, y da la impresión que presenta elongación y desplazamiento caudal del extremo inferior del tallo cerebral, IV ventrículo y amígdalas cerebelosas hacia el canal raquídeo, que sugieren la presencia de una anomalía de la unión craneoespinal compatible con una malformación Chiari tipo II. En la RMN espinal (figura 3) se comprueba la deformidad espinal con curvatura cifótica de la región dorsal, y la presencia de un saco meníngeo abombado hacia el sitio del defecto cutáneo. Presenta, además, hipoplasia de los segmentos caudales de la columna vertebral.
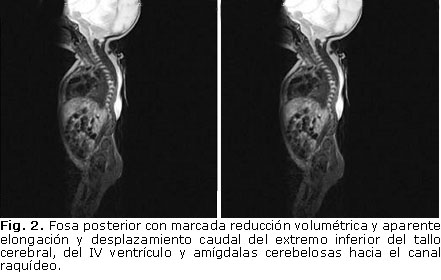
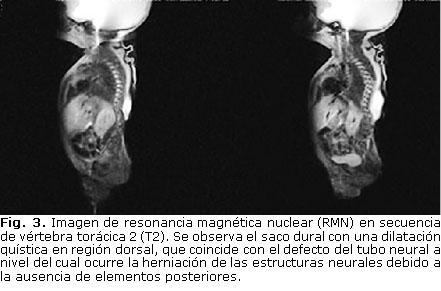
Se lleva al salón de operaciones, se coloca derivación ventrículo-peritoneal y se realiza reparación quirúrgica del defecto del tubo neural. La evolución posoperatoria fue favorable, aunque persistieron los déficit neurológicos preoperatorios. Posteriormente se realiza tomografía axial computarizada (TAC) de 64 cortes para el estudio de la malformación ósea, donde se detecta extenso defecto óseo de los elementos posteriores de la columna vertebral, hemivértebra de la 5ta. lumbar, con agenesia unilateral derecha del sacro y ausencia del cóccix (figura 4).
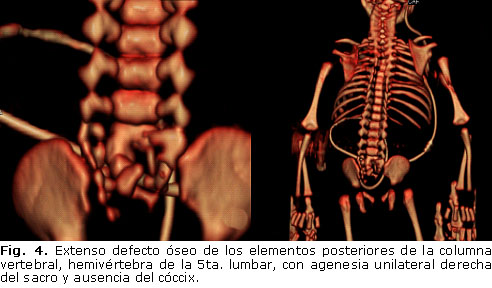
Dentro del estudio se incluyó también una ecocardiografía doppler y una ecografía renal-vesical, con el fin de descartar otras malformaciones orgánicas asociadas. Los resultados de ambos exámenes no revelaron ninguna alteración anatómica significativa a nivel cardíaco ni nefro-urinario. El estudio del cariotipo fue normal.
DISCUSIÓN
En la literatura existen varias teorías sobre el origen del SRC, dentro de las cuales se describen alteraciones de la neurolización primaria o secundaria, trastornos mesodérmicos del extremo caudal de la cuerda embrionaria temprana, y la alteración generalizada de la migración mesodérmica durante el período de estría primitiva, con la fusión de los primordios de los miembros en sus márgenes fibulares, con ausencia o desarrollo incompleto de las estructuras caudales intercurrentes.5,8,9
La incidencia de agenesia lumbosacra es de 1 caso por cada 25 000 nacidos vivos, sin diferencias por sexo, y es más frecuente en los hijos de madres diabéticas (16 % de los casos).7,8,10 Existen varios estudios que analizan esta asociación entre diabetes y SRC. Dentro de sus conclusiones destaca que los hijos de madres con diabetes pregestacional tendrían un riesgo al menos 200 veces mayor de padecer este síndrome, y que de 16 a 22 % de los niños con SRC son hijos de madres diabéticas, todo lo cual lo convertiría en la malformación más característica de la embriopatía diabética.11 Se ha relacionado también con deficiencia de ácido fólico, avitaminosis, administración de insulina durante el embarazo, fiebre materna durante la gestación, administración de sales de litio y exposición a solventes orgánicos.
El análisis del cariotipo en todos los casos es normal (46XX o 46XY), sin embargo, recientes evidencias sugieren que también existe una contribución genética, al menos parcial, para la aparición de agenesia sacra.8,10 La posible participación de un gen "homeobox" (HLXB9), expresado además en el páncreas, pudiera explicar la relación de SRC y la diabetes. Por otra parte, Welch y Aterman describieron cierta susceptibilidad que puede estar relacionada con un inusual haplotipo de antígenos de leucocitos humanos (HLA), aunque estos reportes, sin embargo, son catalogados esporádicos, y se han encontrado gemelos monocigotos discordantes para la agenesia sacra, por lo que hasta la actualidad no se ha establecido ningún patrón de herencia mendeliano.5,8,9,11,12 En este caso no pudieron ser identificados factores predisponentes y el estudio del cariotipo fue normal.
La clínica de esta entidad es variable y está relacionada con la severidad de la anomalía y los procesos malformativos asociados. La inspección de la espalda revela un hueso prominente correspondiente a la última vértebra. La región glútea está achatada con acortamiento del surco interglúteo.5,8 Las anomalías ortopédicas son llamativas, y pueden incluir luxación de caderas, fusión de alas iliacas, e inestabilidad en la unión espino-pelviana.7,9,12,13 También se presentan anomalías de las extremidades inferiores (pies varos, pie zambo, genu recurvatum, defectos de tibia y peroné y fémures hipoplásicos), así como contracciones en flexión de las rodillas con pliegue poplíteo y contracción en flexión de caderas.7-9,12
La asociación con mielomeningocele ocurre en el 30-40 % de los casos, conjuntamente con otras disrrafias ocultas (30 a 50 %), que ocasionan la disfunción motora de la última vértebra sana detectada.7,12 La hidrocefalia que acompaña al mielomeningocele también se ha visto asociada a malformación Chiari tipo II, anomalía que se ha reportado al nacer en el 85-95 % de pacientes con defectos del tubo neural.6,7 Los hallazgos neurológicos varían desde un déficit mínimo a importantes déficits motores y sensitivos de extremidades inferiores, incluyendo debilidad y atrofia de los segmentos inervados por los nervios distales, a las vértebras defectuosas.5,9,14 En nuestro paciente fueron llamativas las malformaciones de extremidades inferiores, además la hidrocefalia extrema asociada al disrrafismo espinal extenso, con defectos motores y sensitivos importantes.
Pueden presentarse además complicaciones adicionales de los sistemas urogenital (agenesia y ectopia renal, uréteres fusionados), gastrointestinal (incontinencia, encopresis y malformación anorrectal en el 46,5 % de casos). Las malformaciones cardiovasculares han sido descritas, pero no son frecuentes (24 %), y se presentan generalmente defectos septales.5,7,9,12,13 En este caso no se revelaron alteraciones anatómicas en otros sistemas, lo que contribuyó a la evolución favorable posoperatoria.
El diagnóstico se basa, para las formas graves, en una ecografía prenatal realizada en el primer trimestre del embarazo. También puede realizarse ultrasonido transvaginal desde la semana 17 de la gestación, aunque el sacro no está bien osificado a finales del primer trimestre y principios del segundo, por lo que muchos casos se diagnostican tardíamente. La imagen característica de la entidad en esta etapa, es un feto con las extremidades inferiores flexionadas en "posición de Buda".7,10,13
La gravedad de la enfermedad se determina mediante el examen del recién nacido por ecografía posnatal e imágenes de rayos x de columna y extremidades, donde se observan las deformidades esqueléticas. Se debe estudiar la columna cervical, y determinar si tiene inestabilidad atlantoaxoidea u otra anormalía congénita a este nivel.12 La RMN completa el estudio, y permite discriminar, además, la presencia de tejido blando dentro del canal espinal, así como alteraciones óseas o nerviosas.5 Se ha descrito en todos los pacientes una médula que termina encima del cuerpo vertebral intacto, y en algunos casos, en forma de cuña, con la porción dorsal más caudal que la ventral, en contraposición al cono medular normal con suave y gradual estrechamiento. Esta forma explicaría el hecho de que en la agenesia sacra el déficit sensitivo ocurre a un nivel más bajo que el motor.2,5,8,10
El tratamiento necesita de un enfoque multidisciplinario para identificar cualquier alteración anatómica y funcional. La reparación quirúrgica precoz del mielomeningocele (dentro de las 24-36 horas del nacimiento) está indicada en todos los casos, y se debe insertar una derivación ventrículo-peritoneal para corregir la hidrocefalia.
La osteotomía supracondílea del fémur y la estabilización de la columna lumbar con la pelvis, ya sea con ortesis o con autoinjertos de la tibia, son tratamientos puestos en práctica para tratar las deformidades ortopédicas.12 La necesidad de obtener un complejo vertebro-pélvico estable es importante para que el paciente se pueda sentar sin ayuda de las manos y logre pararse.5,12
La valoración de los problemas urológicos reviste especial importancia para evitar las infecciones urinarias repetidas, el reflujo, y en consecuencia, la pielonefritis y la hidronefrosis. Se deben realizar periódicamente urocultivos y estudios de la función renal. El tratamiento es solo de soporte, ya que la enfermedad primaria es irreversible.
El pronóstico de los pacientes con agenesia sacra depende fundamentalmente de la severidad de las malformaciones y órganos implicados. En las formas graves, las complicaciones relacionadas con problemas gastrointestinales y renales pueden ocasionar una muerte temprana neonatal; en cambio, los niños que sobreviven, generalmente presentan inteligencia normal.11,14
REFERENCIAS BIBLIOGRÁFICAS
1. Duhamel B. From the mermaid to anal inperforation. The syndrome of caudal regression. Arch Dis Child. 1961;36:152-5.
2. Joshi M, Yadav S. Lumbosacral agenesis. Indian Journal Radiol Imaging. 2011;15(25):1-4.
3. Guille JT, Benavides R, DeAlba CC, Siriram V, Kumar SJ. Lumbosacral agenesis: a new classification correlating spinal deformity and ambulatory potential. J Bone Joint Surg Am. 2002;84A(1):32-8.
4. Phillips WA. Sacral agenesis. The pediatric spine: principles and practice. 2001;1:193-201.
5. Méndez MJ, Cid E, Rodrigo E. Síndrome de regresión caudal. An Esp Pediatr. 1996;44:405-8.
6. Nazar N, Nazar D. Espina Bífida. Revista Medica Hondureña. 1985;53:120-5.
7. Cuevas C. Lumbosacral Agenesis. Pediatric Orthopaedic Research Fellow. 1995 [citado 20 de julio de 2012]. Disponible en: http://gait.aidi.udel.edu/educate/sacragen.htm
8. Araby SJ, Pacheco BC, Medrano SG. Agenesia caudal en una recién nacida. Presentación de un caso. Rev Mex Pediatr. 2004;71:182-5.
9. Joshi M, Yadav S. Lumbosacral agenesis. Musculoskeletal. 2005;15(2):251-4.
10. Bracho V, Tovar J, Rodríguez M, Moreno B. Sirenomelia. Estudio de cinco casos y revisión de la literatura. VITAE Academia Biomédica Digital [serie en Internet]. 2005 [citado 25 de julio de 2012];24. Disponible en: http://www.bioline.org.br/request?va05012
11. Luque MJ, Fernández R, Tuca MJ, Luco M, De Barbieri F, Tapia JL. Síndrome de Regresión Caudal. Caso Clínico. Rev Chil Pediatr. 2010;81(2):148-54.
12. Loera RG, Rodríguez I, Rodríguez R. Delgado CJ, Cruz A. Agenesia lumbosacra. Medicina Universitaria. 2007;9(34):38-41.
13. Vergara HJ, Cardoso C, Rosales ME, Orellana C. Agenesia lumbosacra: tratamiento y propuesta de nueva clasificación. Acta Ortopédica Mexicana. 2005;19(1):6-12.
14. Boulas MM. Recognition of caudal regression syndrome. Adv Neonatal Care. 2009 Apr;9(2):61-9.
Recibido: 16 de octubre de 2012.
Aprobado: 24 de noviembre de 2012.
Lisett Hernández León. Hospital "Abel Santamaría". Km 89, Carretera Central, municipio Pinar del Río. Pinar del Río, Cuba. Correo electrónico: lisett@princesa.pri.sld.cu
