










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Pediatría
versión impresa ISSN 0034-7531
Rev Cubana Pediatr vol.86 no.1 Ciudad de la Habana abr.-jun. 2014
Revista Cubana de Pediatría. 2014; 86(1)
ARTÍCULO ORIGINAL
Evolución de niños con anomalías del tracto urinario y propuesta de interrupción del embarazo
Progress of children with urinary tract anomalies and proposal of pregnancy cessation
Dr. Sandalio Durán Álvarez,I Dra. Rosario Calviac Mendoza,I Dra. Niurka Díaz Zayas,I Dra. Raisa Durán Menéndez,II Lic. Marilyn Pérez Valdés,III Lic. Elvia Luz Sánchez MendozaIII
IHospital Pediátrico Universitario "William Soler". La Habana, Cuba.
IIHospital General Universitario "Enrique Cabrera". La Habana, Cuba.
IIICentro de Investigaciones Clínicas. La Habana, Cuba.
RESUMEN
Introducción: el ultrasonido diagnóstico prenatal realizado en los embarazos normales ha demostrado que por cada 500 embarazos debe aparecer una anomalía importante del tracto urinario. En diferentes situaciones puede sugerirse o recomendarse la interrupción del embarazo, que puede ser aceptado o rechazado por los padres.
Objetivos: comparar el diagnóstico pre y posnatal, y valorar la evolución en 8 pacientes en los que se propuso la interrupción, pero el embarazo continuó.
Resultados: en 6 de los fetos se propuso la interrupción por el diagnóstico de hidronefrosis bilateral; en uno, por quistes renales bilaterales, y en otro por hidronefrosis unilateral y displasia renal multiquística contralateral. En 2 recién nacidos hubo coincidencia total entre el diagnóstico prenatal y el posnatal, en uno con reflujo de alto grado se encontró ureterohidronefrosis bilateral en el estudio prenatal, mientras que en 2 solamente hidronefrosis; un paciente tiene megauréter bilateral no obstructivo, y otro pielectasia bilateral. En el feto que se plantearon los quistes renales bilaterales, el estudio posnatal mostró un doble sistema excretor derecho con el superior obstruido, y reflujo vesicoureteral grado III del inferior con riñón izquierdo normal. Durante el tiempo de seguimiento la conducta médica varió de acuerdo con el diagnóstico posnatal. Al concluir el período de seguimiento, un paciente tiene una enfermedad renal crónica etapa 3, y los 7 restantes tienen función renal conservada.
Conclusiones: la indicación de interrupción del embarazo por el diagnóstico ultrasonográfico prenatal de una anomalía renal o de tracto urinario tiene un margen de error que es necesario seguir estudiando y buscar indicadores de alto riesgo vital, porque los factores predictivos no están bien precisados.
Palabras clave: ultrasonido materno-fetal, anomalías de riñón y tracto urinario, reflujo vesicoureteral, hidronefrosis obstructiva, interrupción del embarazo.
ABSTRACT
Introduction: prenatal ultrasound diagnosis performed in normal pregnancies has shown that one significant urinary tract anomaly occurs per 500 pregnancies. Under different circumstances, termination of pregnancy may be suggested or recommended, which may be accepted or rejected by parents.
Objectives: to compare the pre-and postnatal diagnoses, and to assess the progress in 8 patients who were recommended to terminate their pregnancies, but they rejected this idea.
Results: in 6 cases, the termination of pregnancy was suggested on account of bilateral hydronephrosis diagnosis in their fetuses; in one case due to bilateral renal cysts diagnosis and in the other case due to unilateral hydronephrosis and contralateral multicystic renal dysplasia. There was full agreement between the prenatal and postnatal diagnoses in 2 newborns; in a neonate with high grade reflux, the prenatal study revealed bilateral ureterohydronephrosis whereas this study showed just hydronephrosis for other two fetuses. One patient has non-obstructive bilateral megaureter and the other presents bilateral pyelectasy. The fetus with bilateral renal cysts presented, according to the postnatal study, a double right excretory system, being the upper obstructed and the lower with grade III vesicoureteral reflux, but his left kidney was normal. In the follow-up period, the medical behavior varied according to the postnatal diagnoses. Upon finishing this period, one patient had phase III chronic renal disease and the other seven had preserved renal function.
Conclusions: the indication of termination of pregnancy based on the prenatal ultrasonographic diagnosis of a renal or urinary tract anomaly has an error index that must be further studied, and it is necessary to look for high life risk indicators because the predictive factors are not well detailed.
Keywords: maternal fetal ultrasound, kidney and urinary tract anomalies, vesicoureteral reflux, obstructive hydronephrosis, termination of pregnancy.
INTRODUCCIÓN
Las anomalías congénitas dentro del espectro CAKUT (siglas en inglés de congenital anomalies of the kidney and urinary tract) son la causa más frecuente de enfermedad renal crónica en el niño, y mientras la mayoría de los casos son esporádicos, las anomalías familiares son frecuentes, con una fuerte contribución genética en su origen,1 y motivan frecuentemente consulta prenatal.2 Ocurre 1 anomalía en cada 500 nacimientos, y además de ser la principal causa de insuficiencia renal crónica terminal en Pediatría, predisponen al individuo a la hipertensión arterial y enfermedad cardiovascular en el transcurso de la vida.3 Las anomalías estructurales incluyen agenesia renal, displasia renal multiquística, doble sistema colector, valvas de uretra posterior y anormalidades ureterales.1 Su hallazgo prenatal ha mejorado el pronóstico, y su evaluación requiere un equipo de acceso multidisciplinario que comience antes del nacimiento del niño.4 Su diagnóstico precoz puede permitir que el mejor tratamiento médico o quirúrgico sea implementado tan rápido como sea posible, previniendo, o al menos enlenteciendo, una evolución a la insuficiencia renal crónica.5
En los casos de anomalías severas diagnosticadas tempranamente durante el embarazo, en ocasiones, se sugiere o recomienda la interrupción de este (aborto terapéutico), que puede ser aceptado o rechazado por los padres.4
Aunque en el Hospital Pediátrico Universitario "William Soler" solo se reciben recién nacidos o lactantes en los que prenatalmente se diagnosticaron estas anomalías, y casos que escaparon al diagnóstico prenatal, el objetivo de esta presentación es el análisis evolutivo de los niños, en los que, habiéndose planteado el aborto terapéutico, este fue rechazado por los padres por diferentes motivos, o por lo avanzado de este se decidió continuarlo.
MÉTODOS
Se describen 8 pacientes recibidos en la Consulta de Nefrología del Hospital Pediátrico Universitario "William Soler", y que se logró su seguimiento, nacidos entre el 1º de enero de 2001 y 30 de junio 2013, en los cuales se propuso la interrupción del embarazo, pero el embarazo continuó. En todos los niños se realizó ultrasonido renal y de tracto urinario al llegar a la Consulta de Nefrología, en todos los casos se hizo gammagrafía estática, en 6 de ellos se hizo gammagrafía dinámica, en 6 se realizó uretrocistografía miccional y en 3 urograma excretor. El tiempo de seguimiento varió entre 144 y 5 meses (edad media 51,7 meses). En todos los casos se hizo determinación de creatinina sérica evolutiva y hematogasometría. Dos pacientes fueron sometidos a intervención quirúrgica para la corrección de la anomalía detectada.
Los datos del estudio prenatal fueron obtenidos en el interrogatorio a las madres, excepto en un caso que se revisó la historia clínica de la maternidad.
RESULTADOS
En los 8 pacientes el diagnóstico prenatal se estableció entre las 20 y 26 semanas de gestación (6 fetos de sexo masculino y 2 del femenino). En un feto no se hizo la interrupción por lo avanzado del embarazo, y en 7 por no aceptación de los padres.
En 2 casos (25 %) hubo coincidencia total entre el diagnóstico prenatal y posnatal (pacientes 4 y 6), en 1 con reflujo vesicoureteral (RVU) de alto grado, el estudio prenatal apreció ureterohidronefrosis bilateral (paciente 3), y en los otros 2 casos de RVU solo se encontró dilatación hidronefrótica (pacientes 1 y 8). En un feto (paciente 2) se plantearon quistes renales bilaterales -que no coincidió con el diagnóstico posnatal- ya que después del nacimiento el riñón izquierdo era normal y en el derecho se observó dilatación del polo superior, sin precisarse parénquima. La gammagrafía demostró polo superior afuncional, y la uretrocistografía miccional un RVU grado III del uréter inferior (doble sistema excretor con obstrucción del superior y RVU del inferior). En 2 casos el estudio prenatal reportó hidronefrosis bilateral: en uno el estudio posnatal encontró megauréter no obstructivo bilateral con un riñón congénitamente afectado (paciente 5); y en el otro (paciente 7), solo se observó pielectasia en el estudio posnatal sin afectación en el estudio gammagráfico (tabla 1). En todos, menos en el caso con pielectasia bilateral, los estudios posnatales demostraron afectación de uno o ambos riñones (tabla 2).
Tabla 1. Características pre y posnatales de las alteraciones ultrasonográficas
| Caso | Sexo | Color de la piel | Diagnóstico | Semana de gestación | Diagnóstico posnatal | Tiempo de seguimiento |
| 1 | M | Blanca | Hidronefrosis | 26 | Reflujo vesicoureteral grados IV y V | 144 |
|
2 |
F |
Blanca |
Quistes renales bilaterales |
22 | Doble sistema excretor derecho, superior obstruido, interior RVU grado III, riñón izquierdo normal |
123 |
| 3 | M | Blanca | Ureterohidronefrosis bilateral | 22 | RVU grado V bilateral | 42 |
| 4 | F | Mestiza | Hidronefrosis | 20 | Hidronefrosis obstructiva bilateral | 33 |
| 5 | M | Negra | Hidronefrosis | 24 | Megauréter no obstructivo bilateral | 26 |
|
6 |
M |
Mestiza |
Hidronefrosis derecha, DRMQ izquierda |
22 | Hidronefrosis obstructiva derecha, celda renal izquierda vacía (involución total de los quistes) |
22 |
| 7 | M | Blanca | Hidronefrosis bilateral | 22 | Pielectasia bilateral | 19 |
| 8 | M | Blanca | Hidronefrosis bilateral | 24 | RVU grado V bilateral | 5 |
RVU: reflujo vesicoureteral, DRMQ: displasia renal multiquística
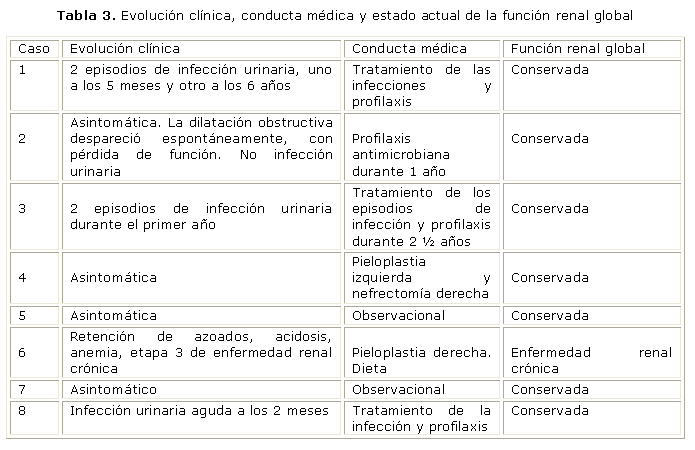
En los pacientes 4 y 6 se hizo tratamiento quirúrgico (pieloplastia izquierda más nefrectomía derecha, y pieloplastia derecha, respectivamente). En los 6 restantes la conducta ha sido el tratamiento de las infecciones, y profilaxis antimicrobiana en los casos 1, 2, 3 y 8; mientras, en los pacientes 5 y 7, solo observación y control clínico imaginológico (tabla 3).
Durante el estudio el único paciente con afectación de la función renal correspondió al niño con displasia renal multiquística (DRMQ) e hidronefrosis obstructiva en el riñón contralateral.
DISCUSIÓN
Hogan y otros, en París2 estudiaron 10 niños, en los que se había propuesto la interrupción del embarazo por las anomalías del tracto urinario detectadas prenatalmente, pero la propuesta fue rechazada por los padres. De estos, 5 murieron en el primer mes de vida; su cifra de creatinina sérica media a los 3 días de nacidos fue 56 µmol/L (rango 25 a 316). Se encontró oligohidramnios en 3 de los pacientes que fallecieron. Los que sobrevivieron, se analizaron a una edad media de 29 meses, y todos tenían creatinina sérica normal con un grado variable de proteinuria (2 de los sobrevivientes también presentaron oligohidramnios). En sus conclusiones plantean que ningún factor predictivo parece tener suficiente especificidad para motivar la propuesta de un aborto terapéutico, y sugieren la necesidad de un estudio con seguimiento prolongado.
Antonsson y otros, en Estocolmo,6 compararon los hallazgos ultrasonográficos y los de la autopsia en 112 casos de interrupciones de embarazo, y encontraron coincidencia total en 45 % de los casos; en 40 % la autopsia confirmó los hallazgos imaginológicos, pero aportó información adicional de importancia clínica. Hubo desacuerdo total en 4 % y desacuerdo parcial en 11 % de los casos. Este estudio incluyó todas las anomalías congénitas, y la coincidencia más baja se obtuvo en las anomalías del sistema nervioso central.
Newbould y otros, en Manchester,7 estudiaron el valor de los hallazgos de la autopsia en 86 lactantes muertos con hallazgos de la secuencia oligohidramnios, con particular referencia a las anomalías del tracto urinario. Hubo 32 con agenesia renal bilateral, 30 con displasia quística bilateral, 8 con agenesia unilateral con displasia quística contralateral, 4 con características histológicas de una alteración con herencia recesiva (2 con disgenesia tubular renal y 2 riñones poliquísticos autosómicos recesivos), 9 presentaban alteraciones menores del tracto urinario y 3 tenían tracto urinario morfológicamente normal.
Hubo 48 lactantes que tenían anomalías distintas a las que produce la secuencia oligohidramnios; más frecuentemente, estas eran anomalías de la asociación VATER esporádica, pero en 4 las anomalías permitieron el reconocimiento de un síndrome hereditario autosómico recesivo (Meckel en 3 casos, y Smith-Lemli-Opitz en 1).
Faure y otros, en Estrasburgo, Francia,8 en un estudio prospectivo de ultrasonido transvaginal entre 5 240 embarazos, identificaron 16 casos de megavejiga fetal precoz. En 15 casos pudieron realizar cariotipo. En 6 casos la megavejiga era aislada, y en los 10 restantes se detectó la asociación con higroma en 5 casos, translucencia nucal en 3, onfalocele en 1 y pielectasia ligera en 1. En 3 casos se constató hiperecogenicidad renal con quistes y oligohidramnios en 2. En el 25 % (4 casos) se encontró anomalía cromosómica: 47,XY+13 (2 casos), 47,XY+18 (1 caso) y 47,XY+21 (1 caso). Solamente sobrevivió 1 caso en este estudio. A esta anomalía se asocia un alto porcentaje de otras malformaciones, y, señalan los autores, la importancia de la evaluación sistemática de enzimas digestivas en el líquido amniótico y orina, para diferenciarla de la disgenesia cloacal.
El factor hepatocito nuclear 1 beta (HNF1-beta) es un factor de transcripción crítico para el desarrollo de riñones y páncreas, y las mutaciones en HNF1-beta conducen a anomalías congénitas de riñón y tracto urinario, atrofia pancreática, diabetes tipo MODY (Maturity Onset Diabetes of the Young) subtipo 5 y agenesia gonadal.9 Heidit y otros, en París,10 estudiaron una cohorte de 377 casos no emparentados con varios fenotipos renales (riñones hiperecogénicos con tamaño no mayor de 3 desviaciones estándar, enfermedad renal multiquística, agenesia renal, hipoplasia renal, displasia quística o nefropatía tubulointersticial hiperuricémica). Encontraron mutación heterocigótica en 75 (19,9 %) casos índices, consistente en una deleción de todo el gen en 42, deleción de un exon en 1 y pequeñas mutaciones en 32. Con este estudio demostraron que la severidad de una enfermedad asociada con mutaciones en HNF1-beta es extremadamente variable (desde insuficiencia renal prenatal, hasta función renal normal en el adulto), y no se corresponde con el fenotipo.
En la hipodisplasia renal se han encontrado mutaciones de los genes PAX2, NHF1-beta, TCF2 y EYA1, que codifican factores críticos para el desarrollo inicial del riñón.4
Madariaga y otros, en París,11 realizaron pesquisa a los genes HNF1-beta y PAX2, en 103 fetos de 91 familias, con CAKUT muy severo, que condujo a la interrupción del embarazo, e identificaron enfermedad causada por mutación de HNF1-beta en 12 casos de 11 familias, así como una mutación de PAX2 en 4 casos no emparentados, y concluyeron que en los casos con CAKUT severo no son raras las mutaciones genéticas.
El RVU se encontró en el 50 % de nuestros pacientes, en 3 de ellos reflujos bilaterales de alto grado, y en una niña asociado a otra anomalía, y la evidencia inicial sugiriendo un origen genético en algunos casos de esta anomalía proviene de los estudios en gemelos, que han demostrado 80-100 % de concordancia en gemelos monocigóticos y solo 30-50 % en los dicigóticos.12 Otras evidencias son la agrupación de casos en familias,13 las diferencias étnicas,14 y el riesgo de aparición de RVU en familiares del caso índice.15,16 En los reportes de familias estudiadas se señalan varios tipos de herencia,17 y en el estudio de Bertoli-Avella el 82 % de los pacientes tienen antecedentes familiares de este tipo de anomalía.18 En la serie de casos que reportamos no se recoge antecedente de anomalías en la familia.
Se ha demostrado que las mutaciones de ROBO2 contribuyen a la patogénesis de RVU/CAKUT en una pequeña proporción de familias.19 Feather y otros20 demostraron ligazón al cromosoma 1p13 para el RVU en forma autosómica dominante, y el estudio realizado por Conte y otros17 sugiere la presencia de varios nuevos locus para el RVU, lo que aporta una nueva evidencia de la heterogeneidad genética de esta anomalía.
Además de los RVU de alto grado -que fue la anomalía más frecuentemente observada en nuestro trabajo, y que el estudio prenatal hacía sospecharlo en 1 de ellos- también se encontró asociado en un doble sistema excretor con obstrucción del sistema superior. Se diagnóstico hidronefrosis por obstrucción ureteropélvica (bilateral en un paciente), y asociada a displasia renal multiquística contralateral en otro; megauréter no obstructivo bilateral en un paciente, y pielectasia bilateral sin afectación renal en otro. El único paciente con evolución a la insuficiencia renal crónica tenía diagnóstico prenatal de DRMQ izquierda e hidronefrosis derecha; los quistes involucionaron antes del nacimiento, los estudios gammagráficos demostraron un monorreno funcional y el riñón hidronefrótico tenía daño prenatal, que, a pesar de la pieloplastia con comprobación gammagráfica de solución de la estenosis ureteropiélica, lo condujo a la enfermedad renal crónica etapa 3 al concluir el estudio. El RVU, diagnóstico más frecuente en esta serie, se considera que es el diagnóstico posnatal entre 9 y 20 % de las hidronefrosis prenatales,21 y en nuestro medio se ha encontrado en 14,7 % de los casos con anomalías del tracto urinario diagnosticadas prenatalmente.22
Después del análisis evolutivo de nuestra pequeña casuística y la breve revisión de la literatura, se considera que la interrupción de embarazo debe ser analizada cuidadosamente por un equipo multidisciplinario, porque muchos de estos casos, si sobreviven, tienen pobre calidad de vida. Aunque no es lo más frecuente, pueden ser hereditarios, por lo que recomendamos el interrogatorio detallado buscando antecedentes familiares de anomalías renales y la pesquisa entre hermanos, mediante ultrasonido renal y de tracto urinario, ya que muchas de estas anomalías pueden cursar asintomáticas durante mucho tiempo.
REFERENCIAS BIBLIOGRÁFICAS
1. Renkema KY, Winyard PJ, Skovorodkin JN, Levtchenko E, Hindryckx A, Jeanpierre C, et al. Novel perspectives for investigating congenital anomalies of the kidney and urinary tract (CAKUT). Nephrol Dial Transplant. 2011;26:3843-51.
2. Hogan J, Dourthe ME, Blondiaux E, Jeanpierre JM, Garel C. Ulinski T. Renal outcome in children with antennal diagnosis of severe CAKUT. Pediatr Nephrol. 2012;27:497-502.
3. Song R, Yosipiv IV. Genetic of congenital anomalies of the kidney and urinary tract. Pediatr Nephrol. 2011;26:353-64.
4. La Scola C, Hewitt I, Pasini A, Pugliese F, Montini G. Postnatal management of congenital bilateral renal hypodysplasia. J Matern Fetal Neonatal Med. 2010;23(suppl 3):97-100.
5. Cauilo VA, Cauilo S, Gargasole C, Chiriacó G, Latini G, Cataldi L, et al. Ultrasound mass screening for congenital anomalies of the kidney and urinary tract. Pediatr Nephrol. 2012;27:949-53.
6. Antonsson P, Sundberg A, Kublickas M, Pilo C, Ghazi S, Westgren M, et al. Correlation between ultrasound an autopsy findings after 2nd trimester terminations of pregnancy. J Perinat Med. 2008;36:59-69.
7. Newbold MJ, Lendon M, Barson AJ. Oligohidramnios sequence: the spectrum of renal malformations. Br J Obstet Gynecol. 1994;101:598-604.
8. Faure R, Kohler M, Gasser B, Muller F, Nisand I. Early fetal megacystis between 11 and 15 weeks of gestation. Ultrasound Obstet Gynecol. 1999;14:402-6.
9. Gómez Ayala AE. MODY diabetes: adult-onset type diabetes in the young. MEDWAVE. 2010;10(2):e4415.
10. Heidit L, Decramer S, Pawtowski A, Morinieri V, Bandin F, Knebelmain B, et al. Spectrum of HNF1B mutations in a large cohort of patients who harbour renal diseases. Clin J Am Soc Nephrol. 2010;5:1079-90.
11. Madariaga A, Moriniére V, Jeanpierre C, Bouvier R, Loget P, Martinovic J, et al, Severe prenatal renal anomalies associated with mutations in HNF1B or PAX2 genes. Clin J Am Soc Nephrol [serie en Internet]. 2013 Mar 28 [citado 28 de junio de 2013];8. Disponible en: http://www.ncbi.nlm.nih.gov/pubmed/23539225
12. Kaefer M, Curran M, Treves ST, Bauer S, Hendren WH, Peters CA, et al. Sibling vesicoureteral reflux in multiple gestation births. Pediatrics. 2000;105:800-4.
13. Tobenkin MI. Hereditary vesicoureteral reflux. South Med J. 1964;57:139-47.
14. King LR. Vesicoureteral reflux: a radiographic sign common to multiple disease. JAMA. 1972;220:854.
15. Kenda RB, Fettick JJ. Vesicoureteral reflux and renal scars in asymptomatic siblings of children with reflux. Arch Dis Child. 1992;67:506-8.
16. Parekh DJ, Pope JC, Adams MC, Brock JW. Outcome of sibling vesicoureteral reflux. J Urol. 2002;167:283-4.
17. Conte ML, Bertoli-Avella AM, de Graaf BM, Punzo F, Lama G, La Manna A, et al. A genome search for primary vesicoureteral reflux shows further evidence for genetic heterogeneity. Pediatr Nephrol. 2008;23:587-95.
18. Bertoli-Avella AM, Conte ML, Punzo F, de Graaf BM, Lama G, La Manna A, et al. ROBO2 gene variants are associated with familial vesicoureteral reflux. J Am Soc Nephrol. 2008;19:825-31.
19. Lu W, van Eerle AM, Fan X, Quintero-Rivera F, Kulkarni S, Ferguson H, et al. Disruption of ROBO2 is associated with urinary tract anomalies and confers risk of vesicoureteral reflux. Am J Hum Genet. 2007;80:616-32.
20. Feather SA, Malcolm S, Woolf AS, Wright V, Blaydon D, Reid CJ, et al. Primary, nonsyndromic vesicoureteral reflux and its nephropathy is genetically heterogeneous, with a locus on chromosome 1. Am J Hum Genet. 2000;66:1420-5.
21. Sargent MA. What is the normal prevalence of vesicoureteral reflux? Pediatr Radiol. 2000;30:587-93.
22. Durán Álvarez S, Betancourt González U, Hernández Hernández JS, Campañá Cobas NG, González Pérez O. Diagnósticos posnatales de anomalías del tracto urinario detectadas mediante ultrasonido materno-fetal. Rev Cubana Pediatr [serie en Internet]. 2004 [citado 4 de agosto de 2013];76(4). Disponible en: http://scielo.sld.cu/scielo.php?script=sci_arttext&pid=S0034-75312004000400003&lng=es&nrm=iso&tlng=es
Recibido: 1º de agosto de 2013.
Aprobado: 4 de septiembre de 2013.
Sandalio Durán Álvarez. Hospital Pediátrico Universitario "William Soler". San Francisco # 10 112, Reparto Altahabana, municipio Boyeros. La Habana, Cuba. Correo electrónico: sduran@infomed.sld.cu

{kind=link}
{kind=link}