










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Pediatría
versión impresa ISSN 0034-7531
Rev Cubana Pediatr vol.86 no.1 Ciudad de la Habana abr.-jun. 2014
Revista Cubana de Pediatría. 2014; 86(1)
PRESENTACIÓN DE CASO
Caso atípico de osteoporosis idiopática juvenil
Atypical case of juvenile idiopathic osteoporosis
Dr. Iván Hernández García,I Dra. Mirelkys Bustamante Teijido,I MSc. Dra. Mérida Rodríguez Orta,II Dra. Silvia León Pérez, II Dra. Alina García GarcíaI
IHospital Pediátrico Docente "William Soler". La Habana, Cuba.
IIÁrea de Salud de Nueva Paz. Mayabeque, Cuba.
RESUMEN
La osteoporosis idiopática juvenil es una enfermedad que aparece en niños y adolescentes, autolimitada, caracterizada por osteoporosis axial y de extremidades, en ocasiones asociada a colapso vertebral, lo que resulta finalmente en cifosis torácica. No se tiene conocimiento, hasta la fecha, de su asociación a alteraciones de la dentición. Se presenta el caso de un adolescente masculino, con historia de retraso en el cambio a la dentición permanente y osteopenia, valorado por varias especialidades médicas como: Medicina Interna, Estomatología, Ortopedia, Endocrinología y Nefrología. Se plantea el diagnóstico de osteoporosis idiopática juvenil, y se descartan, a partir del cuadro sintomatológico y los numerosos complementarios realizados, varias enfermedades hereditarias y adquiridas, entre ellas, la osteogénesis imperfecta tardía y el hiperparatiroidismo. Se destacan en este caso la presencia de alteraciones en la dentición y la poca afectación vertebral.
Palabras clave: osteoporosis, osteoporosis idiopática juvenil, alteraciones en la dentición, osteopenia.
ABSTRACT
Juvenile idiopathic osteoporosis is a disease seen in children and adolescents, it is self-limited, characterized by axial and limb osteoporosis, occasionally related to vertebral collapse, which finally resulted in thoracic kyphosis. To date, the association between this disease and dental impairments is not known. Here is the case of a male adolescent, with history of delayed change to permanent dentition and osteopenia; he was evaluated by several medical specialists as internal medicine, odontology, orthopedics, endocrinology and nephrology. He was diagnosed as juvenile idiopathic osteoporosis case, and on the basis of the symptom picture and the numerous supplementary tests, it was possible to rule out several hereditary and acquired illnesses such as late imperfect osteogenesis and hyperparathyroidism. This case stressed the presence of dentition impairments and little vertebral effect.
Keywords: osteoporosis, juvenile idiopathic osteoporosis, dentition impairments, osteopenia.
INTRODUCCIÓN
La osteoporosis se produce cuando hay un desequilibrio entre la formación y la resorción ósea, aumento de la resorción, o déficit en la formación del hueso. Hay un déficit tanto de la matriz ósea como del calcio (Ca+) y otros minerales que la forman. El hueso es cualitativamente normal, pero cuantitativamente anormal. La OMS considera que hay osteoporosis sobre la base de la densidad mineral del hueso, medida por densitometría, y comparada con una población de referencia (T-Score £ 2,5).1 En resumen, se puede afirmar que existe osteoporosis cuando la pérdida ósea es mayor que la esperada para una persona de edad, sexo y raza determinados, o cuando hay un déficit estructural del hueso y se producen fracturas recurrentes.1
La osteoporosis se asocia, sobre todo, con el anciano y con las mujeres en etapa posmenopáusica, sin embargo puede verse en niños y jóvenes, asociada a otras enfermedades. Cuando no aparece secundariamente y se presenta en etapas tempranas de la vida, es pertinente el diagnóstico de osteoporosis idiopática juvenil (OIJ).
La OIJ, entidad probablemente autosómica recesiva, delineada por Dent y Friedman en 1965, está aún insuficientemente estudiada, razón por la que no se conoce con precisión la frecuencia y aspectos elementales de su genética molecular, así como el mecanismo fisiopatológico por el que se produce la enfermedad.2,3 La OIJ es autolimitada, y se caracteriza por osteoporosis axial, en ocasiones, asociada a colapso vertebral, lo que condiciona cifosis torácica. Las extremidades también pueden resultar afectadas con cortezas finas y líneas de fractura más típicamente en metáfisis de rodillas y tobillos.3 El diagnóstico diferencial con otras enfermedades hereditarias o adquiridas puede resultar difícil, como en el caso de la osteogénesis imperfecta tardía (OIT). El objetivo del presente trabajo es dar a conocer un paciente con una presentación atípica de la enfermedad y las dificultades que entraña su diagnóstico.
PRESENTACIÓN DEL CASO
Adolescente de 18 años, hijo de padres no consanguíneos, de sexo masculino, atendido en Consulta de Genética por presentar osteopenia generalizada. El paciente fue atendido inicialmente en las Consultas de Estomatología y Medicina Interna, por no haber completado el cambio de la dentición, especialistas que detectan osteoporosis (T-Score< 2,5) y lo envían a consulta de Endocrinología, y esta, a su vez, a la de Nefrología, por detectar en los estudios practicados hipercalciuria con hiperfosfaturia. Finalmente es enviado a Consulta de Genética para valorar posibilidades diagnósticas.
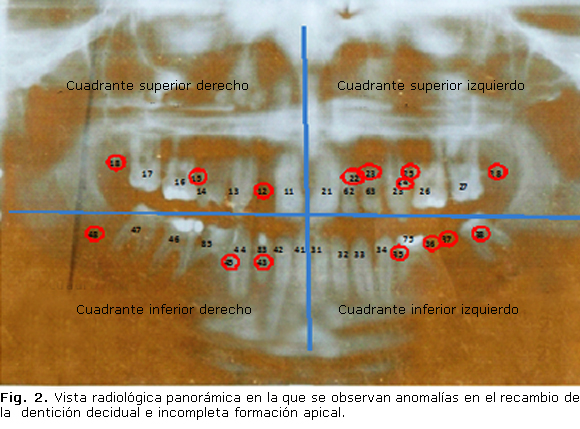
Al examen físico se encuentra talla de 168 cm, peso de 47,5 kg (peso/talla entre 3er y 10mo percentil, y talla/edad en el 50ta percentil), de acuerdo con las tablas de crecimiento y desarrollo de la población cubana. Es un paciente longilíneo, de apariencia delgada. Su desarrollo puberal es adecuado para la edad, estadio IV de Tanner. No se encontró dismorfismo facial. El tiroides no es visible ni palpable, ni tampoco hay deformidades óseas. Se encontró ausencia de varias piezas dentales, porque aún no habían brotado, así como persistencia de dentición decidual (figuras 1 y 2).
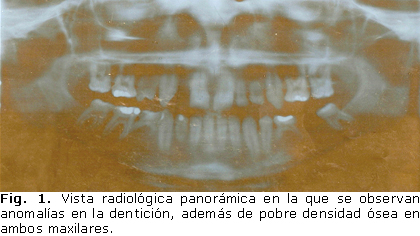
Entre los análisis realizados por las diferentes especialidades, se detectó calcio (Ca+) en sangre de 2,30 mmol/L (valores normales 2,02-2,63) y fósforo (P+) con valores de 1,23 mmol/L (valores normales 1,13-1,61) y aumento en la excreción de ambos minerales en orina, Ca+ 6,3 mmol/kg/24 horas (valores normales 1,3-3,8) y P+ 21,9 mmol/kg/24 horas (valores normales 4,8-14,5). El Ca+ iónico determinado por gasometría en sangre capilar también estaba dentro de límites normales (1,210 mmol/L). Estos análisis fueron repetidos con dieta fija y se obtuvieron similares resultados
(tabla 1).

Otros hallazgos relevantes fueron varias imágenes ecolúcidas en el polo inferior de los lóbulos tiroideos de hasta 9 mm, interpretadas como quistes coloides y adenopatías de pequeño tamaño, de aspecto inflamatorio, de ambas cadenas ganglionares del cuello. También se observó un discreto aumento de las proteínas totales (88 g/L). Los valores de hormona paratiroidea (PTH) y fosfatasa alcalina (FA) fueron normales (tabla 2). Las pruebas metabólicas en orina no mostraron indicios de aminoaciduria generalizada o glucosuria.

La radiografía panorámica (figura 2) mostró en el cuadrante superior derecho (maxilar superior), la presencia del incisivo central superior derecho (11), canino (13), primer premolar (14), primer molar (16) y segundo molar (17), todos permanentes; pero se observó ausencia del incisivo lateral (12), del segundo premolar (15) y del tercer molar (18). En el cuadrante superior izquierdo se observó el incisivo central superior (21), canino (23), primer molar (26) y segundo molar (27), todos permanentes; y, además, reflejó la presencia de dientes pertenecientes a la dentición temporal, como el incisivo lateral (62) y el canino (63). Ausentes (oligodoncia) se encuentran el primer premolar (24) y segundo premolar (25), además del tercer molar (28).
En el cuadrante inferior izquierdo se encuentran presentes el incisivo central (31), el incisivo lateral (32), el canino (33), el primer premolar (34) y el tercer molar (38), todos permanentes, además del segundo molar temporal (75); pero existe ausencia del segundo premolar (35), del primer molar (36) y del segundo molar permanentes (37). En el cuadrante inferior derecho está presente el incisivo central (41), incisivo lateral (42), así como el primer premolar (44), primer molar (46) y segundo molar permanentes (47), y además el canino temporal (83) y el segundo molar temporal (85). Existe ausencia del segundo premolar inferior derecho (45) y del tercer molar permanente (48).
En la dentición permanente se pudo observar la incompleta formación apical de los dientes presentes; además, existe oligodoncia de 12, 15, 18, 22, 23, 24, 25, 28, 35, 36, 37, 38, 43, 45 y 48.
DISCUSIÓN
De acuerdo con el balance fosfocálcico que presenta este paciente se pudo descartar, a partir del cuadro clínico, los estudios radiológicos y hormonales, la presentación de osteoporosis con hipercalciuria en la hipercalciuria absortiva idiopática y la asociada al incremento del 1,25 dihidrocolecalciferol, secundario a enfermedad granulomatosa y a enfermedades con aumento del recambio óseo, como la enfermedad de Paget, el hiperparatiroidismo y el hipertiroidismo (tabla 2). Estas entidades, además, se acompañan con frecuencia de cálculos renales o nefrocalcinosis. Asimismo, como muestra la tabla 1, el paciente presentaba valores en sangre de Ca+ y P+ normales con Ca+ y P+ elevados en orina, por lo cual puede descartarse un hiperparatiroidismo. También en esta entidad encontramos elevación de la FA debido al aumento de la resorción ósea y valores de PTH elevados (hiperparatiroidismo primario), lo que no se detectó en este paciente (tabla 2). El hiperparatiroidismo secundario también cursa con elevación secundaria de Ca+ y P+ en sangre.
Los niveles séricos de Ca+ y P+, electroforesis de proteínas séricas, eritrosedimentación y la FA sérica suelen ser normales en la osteoporosis primaria, lo cual concuerda con este caso. La OIJ en el varón joven puede acompañarse también de hipercalciuria (0,1 mmol/kg/24 horas, o 4 mg/kg/24 horas). En el paciente los niveles de excreción de Ca+ se encontraban alrededor de 0,103 mmol/kg/24 horas, cifras similares a las de los pacientes hipercalciuricos, quienes, sin embargo, no tienen una excreción tan marcada de P+ como en este caso (0,378 mmol/kg/24 horas), y además, padecen de cálculos a repetición y/o hematuria, lo que no se ha constatado en este caso, al que tampoco se encontró signo alguno de afectación tubular (tabla 2).4 Esta última afectación se observa en los pacientes hipercalciúricos, acompañada de acidosis metabólica crónica y presencia o no de depleción de potasio, nefrocalcinosis y osteomalacia o raquitismo.
En el caso descrito no se recoge el antecedente de terapia medicamentosa con anticonvulsivantes o medicamentos esteroideos, no existe clínica de enfermedad hematológica u ósea de carácter tumoral, enfermedad gastrointestinal, hepática, malnutrición o nutrición parenteral, entre otras. Se descartan, por el interrogatorio y el cuadro clínico, varias enfermedades que cursan con osteoporosis generalizada, como el mieloma de células plasmáticas, la diabetes mellitus, la inmovilización prolongada, los estados de inmunodeficiencia, la enfermedad de Gaucher, la glucogenosis y otras.
La OIT es muy difícil de distinguir de la OIJ, pero en el primer caso el paciente presenta, además de la osteoporosis, escleras azules y fracturas a repetición, sobre todo, de huesos largos. Tampoco se recoge en la OI una excreción por encima de niveles fisiológicos de Ca+ y P+ en orina. La OIJ se clasifica como una osteoporosis de bajo recambio o remodelamiento óseo, lo cual se corresponde con los valores de FA encontrados (tabla 1). Las extremidades también pueden resultar afectadas con cortezas finas y líneas de fractura más típicamente en metáfisis de rodillas y tobillos, aunque este hallazgo distintivo entre ambas entidades no lo hemos constatado aún en este paciente.
En todo varón, sobre todo si es joven, debe investigarse un posible hipogonadismo, para poder descartar una osteoporosis secundaria a un déficit de los esteroides sexuales implicados también en el mantenimiento de la masa ósea. Las determinaciones hechas en este paciente fueron normales (tabla 2).5-7
Otra de las hormonas relacionadas con el metabolismo óseo y fosfocálcico es la calcitonina, hormona encargada de fijar el Ca+ en el hueso. La calcitonina es una hormona polipeptídica con muchas variantes, que se obtienen por empalme alternativo a partir de un mismo transcripto primario (Jacobs y otros, 1981). Está demostrado que existen mutaciones en el gen de la calcitonina que producen osteoporosis, aunque no se han asociado a defectos en la dentición.8 Gorlin y otros, en 1980, describen la completa ausencia de dentición permanente en un paciente, y concluyen que podría tratarse de un una entidad autosómica recesiva con entrada (# 206780) en el catálogo de síndromes de McKusic.8 La literatura recoge otros síndromes con afectación de la dentición permanente y afectación renal, aunque no asociada a osteoporosis ni a la excreción elevada de Ca+ y P+ en orina.9,10
En la amelogénesis imperfecta y calcificación renal, entidad autosómica recesiva, los síntomas iniciales están asociados a enuresis y problemas dentales, sobre todo, oscurecimiento de los dientes. Los incisivos centrales irrumpen a los 7 u 8 años, y se oscurecen con la edad, y son amarillo-marrón. Los restantes fallan en emerger o están faltos de esmalte. Los estudios renales muestran nefrocalcinosis y defecto tubular.9 Forsman y otros localizaron un gen asociado solo con oligodontia.10 Vastardis y otros, en 1996, reportaron una familia con patrón de herencia autosómico dominante que presentaba dentición primaria normal, pero agenesia de algunos de los dientes permanentes, incluyendo los primeros premolares y todos los segundos premolares y terceros molares de las arcadas maxilares y mandibulares. El gen fue mapeado en la región 4p16, y fue demostrada una mutación con pérdida de sentido en el gen Msh homeobox 1 (MSX 1).10 Ninguno de los casos presentaba osteoporosis.
La presentación de la osteoporosis en el niño o el adolescente es rara. El diagnóstico de OIJ requiere descartar numerosas enfermedades sistémicas y el concurso de varios especialistas. Es necesario un estudio de laboratorio acucioso. En la literatura consultada no se encontró la presentación de OIJ con alteraciones en la dentición.
REFERENCIAS BIBLIOGRÁFICAS
1. Michael Lewiecki E. Managing osteoporosis: Challenges and strategies. Cleveland Clinic Journal of Medicine. 2009;76(8):457-66.
2. McKusic VA. Osteoporosis Idiopática Juvenil. En: Online Mendelian Inheritance in Man. Catálogo en línea [homepage en Internet] [citado 19 de abril de 2012]. Disponible en: http://omim.org/entry/259750
3. Castriota-Scanderbeg A, Dallapiccola B. Generalized Skeletal Abnormalities. In: Abnormal Skeletal Phenotypes: From Simple Signs to Complex Diagnose. Berlin-Heidelberg: Springer-Verlag; 2005. p. 501-10.
4. Stechman MJ, Loh NY, Thakker RV. Genetic causes of hypercalciuric nephrolithiasis. Pediatr Nephrol. 2009;24(12):2321-32.
5. Nuti R, Martini G, Merlotti D, De Paola V, Valleggi F, Gennari L. Bone metabolism in men: role of aromatase activity. Endocrinol Invest. 2007;30:18-23.
6. Ashida K, Akehi Y, Kudo T, Yanase T. Bone and Men's Health. The role of androgens in bone metabolism. Clin Calcium. 2010 Feb;20(2):165-73.
7. Gorlin RJ, Herman NG, Moss SJ. Complete absence of the permanent dentition: an autosomal recessive disorder (Letter). Am J Med Genet. 1980;5:207-9.
8. Lubinsky M, Angle C, Marsh PW. Syndrome of amelogenesis imperfecta, nephrocalcinosis, impaired renal concentration, and possible abnormality of calcium metabolism. Am J Med Genet. 1985;20:233-43.
9. Forsman K, Lind L, Backman B. Localization of a gene for autosomal dominant amelogenesis imperfecta (ADAI) to chromosome 4q. Hum Molec Genet. 1994;3:1621-5.
10. Vastardis H, Karimbux N, Guthua SW, Seidman JG, Seidman CE. A human MSX1 homeodomain missense mutation causes selective tooth agenesis. Nature Genetics. 1996;13:417-21.
Recibido: 30 de mayo de 2013.
Aprobado: 9 de septiembre de 2013.
Iván Hernández García. Hospital Pediátrico Docente "William Soler". San Francisco # 10 112, Reparto Altahabana, municipio Boyeros. La Habana, Cuba.
Correo electrónico: ivan.hernandez@infomed.sld.cu
