










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Pediatría
versión impresa ISSN 0034-7531
Rev Cubana Pediatr vol.86 no.3 Ciudad de la Habana jul.-set. 2014
ARTÍCULO ORIGINAL
Poliposis adenomatosa familiar en niños cubanos
Familial adenomatous polyposis found in Cuban children
Dr. Tulio Antonio Amaya Sorto, MSc. Elsa García Bacallao, Dra. Sacha Lazo del Vallín, MSc. Licet González Fabián, Dra. Marianny Torres Fernández, MSc. Nélcido Luis Sánchez García
Instituto de Gastroenterología. La Habana, Cuba.
RESUMEN
Introducción: la poliposis adenomatosa familiar es una enfermedad autosómica dominante con evolución al cáncer colorrectal.
Objetivo: caracterizar a los niños cubanos con poliposis adenomatosa familiar.
Métodos: se realizó un estudio, descriptivo, prospectivo de serie de casos, atendidos en el Instituto de Gastroenterología de Cuba, durante el periodo comprendido entre febrero de 2011 y mayo de 2013. Se incluyeron 15 niños, en los cuales se había establecido el diagnóstico de poliposis por colonoscopia, con confirmación histológica de adenomas. A todos se les realizó endoscopia del tracto digestivo superior, ultrasonografía de abdomen superior, ortopantomografía, survey óseo, tránsito intestinal, consulta de oftalmología y neurología.
Resultados: el 60,0 % fue del sexo masculino y el 60,0 % de color de piel blanca. La pesquisa de los pacientes asintomáticos y el sangrado rectal fueron los motivos de consulta más frecuentes (40,0 % respectivamente). Predominó la forma florida de la enfermedad, y la displasia de bajo grado se observó en el 73,3 %. El 26,7 % tuvo pólipos en el estómago, y fue la localización más observada. La manifestación extraintestinal más frecuente fue la hipertrofia congénita del epitelio retiniano (73,3 %), seguida por los dientes supernumerarios y los quistes dentígenos. Al analizarlo por grupos de edades, entre 10 y 18 años, al 40,0 % ya se les había realizado colectomía.
Conclusiones: la mayoría de los pacientes estudiados tenían antecedentes familiares de la enfermedad, la pesquisa familiar y el sangrado rectal fueron los principales motivos de estudio. Todos presentaron la forma florida, y en su gran mayoría, displasia de bajo grado en el momento del diagnóstico. Los pólipos extracolónicos se presentaron con mayor frecuencia en el estómago, y la manifestación extraintestinal más frecuente fue la hipertrofia congénita del epitelio retiniano. La mayoría de los pacientes no se habían realizado colectomía en el momento del estudio.
Palabras clave: poliposis adenomatosa familiar.
ABSTRACT
Introduction: familial adenomatous polyposis is a dominant autosomal disease that evolves into colorectal cancer.
Objective: to characterize the Cuban children with familial adenomatous polyposis.
Methods: a prospective, descriptive case series study, who were seen at the Institute of Gastroenterology from February 2011 through May 2013. Fifteen children were diagnosed as adenomatous polyposis patients, based on colonoscopy and histological confirmation, respectively. All of them were performed upper digestive tract endoscopy, upper stomach ultrasonography, ortopantomography, bone survey, intestinal transit test, and ophthalmological and neurological exams.
Results: sixty percent were males and 60 % were Caucasians. Screening of asymptomatic patients and the rectal bleeding were the main causes of medical consultation (40 %, respectively). Full manifested disease prevailed and low-grade dysplasia was observed in 73.3 % of cases. In the study group, 26.7 % presented with polyps in their stomach, being this location the most observed one. The most common extraintestinal manifestation was congenital hypertrophy of the retinal epithelium (73.3 %) followed by supernumerary teeth and dentigerous cysts. On analyzing the age groups, 40 % of the 10 to 18 years old children had already undergone colectomy.
Conclusions: Most of the studied patients had family histories of the disease; the family screening and the rectal bleeding were the main reasons for the study. All of them presented with the fully manifested form of the disease and the vast majority had low grade dysplasia at the time of diagnosis. The polyps located out of the colon were more frequently found in the stomach and the most common extraintesinal manifestation was congenital hypertrophy of the retinal epithelium. The majority of the patients had not undergone colectomy at the time of study.
Keywords: familial adenomatous polyposis.
INTRODUCCIÓN
El pólipo colorrectal se define como protrusión de la mucosa hacia la luz intestinal.1 La poliposis adenomatosa familiar (PAF) es una enfermedad que se hereda de forma autosómica dominante, con 80 a 100 % de penetrancia y expresión fenotípica variable, debido a mutaciones germinales del gen adenomatous polyposis colic (APC), caracterizada por la presencia de numerosos pólipos, habitualmente más de 100 en el colon y/o el recto, que originan la variable denominada clásica o florida.1-5 En algunos casos se trasmite de forma autosómica recesiva, por mutaciones en el gen MUTYH, y originan la variable denominada poliposis adenomatosa familiar atenuada (PAFA), que se caracteriza por presentar entre 10 y 100 pólipos colorrectales, que muestran con frecuencia un patrón de crecimiento plano en lugar de polipoide, con tendencia a agruparse en el colon proximal.1,6 Si no se reseca el colon, el desarrollo de cáncer de colon es casi inevitable.
Los pacientes suelen estar asintomáticos hasta la pubertad, cuando pueden comenzar a aparecer los pólipos en el colon, los que, rara vez aparecen durante la primera década de la vida.1 En los niños, la presencia de pólipos implica la resección endoscópica y/o quirúrgica de estos, pero su seguimiento evolutivo no siempre es el adecuado, tal vez confiando en la benignidad de la lesión y en la evolución poco frecuente al cáncer de colon en la infancia, lo cual no implica que estén exentos de riesgos.1,2
La prevalencia estimada es muy variable, oscila entre de 1:5 000 a 1:20 000 por habitantes.1-5 La edad de inicio de la pesquisa debe ser alrededor de los 12 años, cuando ya comienzan a aparecer los adenomas, y así, establecer un esquema de seguimiento precoz, teniendo en cuenta que se han reportado pacientes con diferentes grados de displasia, e incluso, la aparición de cáncer colorrectal (CCR) aun en la edad pediátrica.1,7,8
La mayoría de los casos con PAF se diagnostican a punto de partida de un estudio de pesquisa familiar siendo pacientes asintomáticos, no obstante las manifestaciones clínicas más frecuentes son: la diarrea crónica, el sangrado rectal, el edema secundario a la enteropatía perdedora de proteínas, la palidez cutánea mucosa secundaria a anemia crónica, así como el dolor abdominal.1,9
En 30 a 100 % de los pacientes pueden encontrarse pólipos en el estómago, que la mayoría son hiperplásicos, de las glándulas fúndicas, que pueden aparecer en la primera década de la vida, incluso, antes del desarrollo de otros adenomas intestinales. Los adenomas gástricos son poco frecuentes, y pueden estar en relación con el desarrollo de adenocarcinoma de estómago.10,11 Los adenomas duodenales aparecen entre 60 y 90 % de los pacientes con PAF, y la incidencia aumenta con la edad, con cierta tendencia a su localización periampular.12,13
La PAF se asocia frecuentemente con manifestaciones extraintestinales como son: hipertrofia congénita del epitelio retiniano pigmentario (HCERP), osteomas, quistes epidermoides, dientes supernumerarios, tumores desmoides, adenoma de glándulas suprarrenales, quistes dentígenos, cáncer de tiroides, cáncer de páncreas, hepatoblastoma y tumores del sistema nervioso central.1,14,15
El objetivo del estudio es caracterizar a los niños cubanos con PAF atendidos en el Instituto de Gastroenterología.
MÉTODOS
Se realizó un estudio, descriptivo, prospectivo de serie de casos, de 15 pacientes en edades pediátricas con el diagnóstico de PAF, atendidos en el Instituto de Gastroenterología de La Habana, Cuba, durante el periodo comprendido entre febrero del año 2011 y mayo del año 2013.
Se establecieron para la realización del estudio como criterios de inclusión a aquellos pacientes en los cuales se había establecido el diagnóstico de poliposis por colonoscopia, con confirmación histológica de adenomas, que accedieron a participar en la investigación, además del consentimiento del padre, madre o tutor; y como criterios de exclusión, aquellos pacientes con los criterios diagnósticos señalados, pero que se negaron a participar en el estudio, o que presentaron alguna contraindicación para la realización de los estudios previstos. A todos, para su diagnóstico, se les realizó una colonoscopia, hasta llegar a visualizar toda la mucosa del colon, y se observaron minuciosamente todas las características de esta, se realizó polipectomía endoscópica para recuperar el o los pólipos, o se tomó muestra para biopsia de estos, a las que se les realizó estudio histológico para precisar la certeza de tratarse de pólipos adenomatosos.
Desde el punto de vista endoscópico se definió como forma florida a los casos que tuvieron 100 o más pólipos localizados en todo el colon, y forma atenuada a los que tenían menos de 100 con localización preferiblemente en colon derecho. A todos se les hizo endoscopia del tracto digestivo superior, para definir la presencia de pólipos; y si estaban presentes, se les realizó polipectomía endoscópica, o se tomó muestra para biopsia en los casos en que esta no fue posible, a las que se les realizó estudio histológico.
A todos los pacientes se les realizó ultrasonografía de abdomen superior para determinar lesiones en el hígado, el páncreas, las glándulas suprarrenales, y además, ultrasonido de tiroides. Igualmente se les realizó ortopantomografía para determinar la presencia de quistes dentígenos, osteomas mandibulares y dientes supernumerarios. El survey óseo se hizo para evaluar la presencia de osteomas y exostosis. Se realizó el estudio radiográfico de tránsito intestinal para determinar la presencia de pólipos en la mucosa del intestino delgado. Se evaluaron también a los pacientes en la consulta de Oftalmología, con el fin de determinar la presencia de HCERP.
Todos los pacientes fueron examinados por el neurólogo para definir signos incipientes de lesión expansiva del sistema nervioso central, y en caso de encontrar alteraciones, estuvo indicada la realización de tomografía axial computarizada. Todas estas variables se analizaron de forma dicotómica como presentes o ausentes.
Para el análisis descriptivo de los datos se utilizaron medidas de frecuencia para variables cualitativas (frecuencias absolutas y relativas). Este estudio fue aprobado por el Consejo Científico y el Comité de Ética del Instituto de Gastroenterología.
RESULTADOS
En la investigación se estudiaron 15 pacientes con el diagnóstico de PAF. Las características generales de estos se exponen en la tabla 1. En el estudio el 60,0 % de los pacientes fueron del sexo masculino, así como el 60,0 % tuvo color de piel blanca. La totalidad de los pacientes presentó antecedentes familiares de PAF.
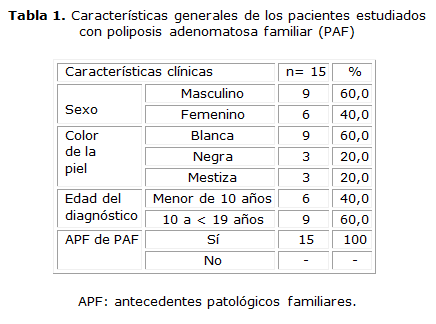
La pesquisa de los pacientes asintomáticos fue el motivo de consulta más frecuente (40 %); no obstante, la manifestación clínica más frecuentemente reportada en los pacientes con PAF fue el sangrado rectal (40 %) seguido por el dolor abdominal (13,3 %) y la diarrea crónica (6,7 %). Todos los pacientes presentaron la forma florida de la enfermedad.
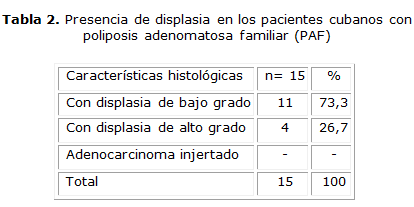
El 73,3 % los pacientes presentaron displasia de bajo grado. Se encontraron 4 pacientes (26,7 %) con displasia de alto grado, y no existió adenocarcinoma injertado como hallazgo histológico (tabla 2).
El 26,7 % tuvo pólipos en el estómago, localización que fue la más frecuente, y todos hiperplásicos desde el punto de vista histológico. El 13,3 % tuvo pólipos en el duodeno (adenomatosos desde el punto de vista histológico), y 1 de ellos con degeneración neoplásica (tabla 3). Hubo 2 pacientes que presentaron pólipos en estómago y duodeno; 1 de ellos con presencia de estos en el estómago, que además los presentó en íleon diagnosticado por tránsito intestinal. Ningún paciente presentó pólipo en el yeyuno. Cuando se analizaron en general los pólipos extracolónicos, estuvieron presentes en el 33,3 % de los pacientes.

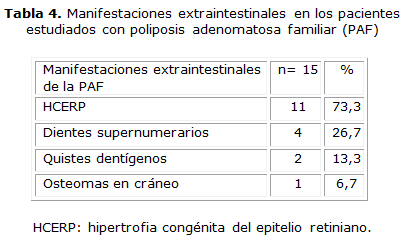
La manifestación extraintestinal más frecuente fue la HCERP (73,3 %). Mediante la realización de la ortopantomografía, los dientes supernumerarios y los quistes dentígenos se observaron en el 26,7 y 13,3 % respectivamente (tabla 4).
Al 26,7 % (4/15) de los pacientes estudiados se les realizó una colectomía total, al determinar la presencia de displasia de alto grado en el estudio histológico de los pólipos; y la colectomía subtotal se realizó en el 6,7 % de los pacientes, con la variante anastomosis ileorectal en el 20,0 %. En el 73,3 % no se ha realizado la colectomía por no cumplir aún con los criterios quirúrgicos (tabla 5).
DISCUSIÓN
Se conoce que los pólipos aparecen con frecuencia alrededor de los 15 años, aunque las manifestaciones clínicas aparecen más tardíamente;1,7,8 sin embargo, más recientemente Erdman y otros7 y Saab y otros,8 sugieren que la edad de inicio de la pesquisa sea alrededor de los 12 años, cuando ya comienzan a aparecer los adenomas, y así establecer un esquema de seguimiento precoz, teniendo en cuenta que se han reportado pacientes con diferentes grados de displasia, e incluso, la aparición de CCR aún en la edad pediátrica.
En el estudio realizado por Alfaro y otros,16 en 243 pacientes con PAF, el 53,5 % fueron del sexo masculino resultado, que coincide con el nuestro; por otro lado, en el trabajo realizado por Björk 17 acerca de la epidemiología de la PAF en Suecia, concluye que el inicio de los síntomas y la aparición del CCR es más temprano en las pacientes femeninas que en los del sexo masculino.
La PAF es una enfermedad hereditaria debido a mutaciones germinales del gen APC, localizado en el cromosoma 5, la cual se precisa en el 85 % de las familias con PAF y en el 20-30 % de los casos de PAFA;1-5,7 sin embargo, el 10-20 % de los casos de PAF clásica y 30 % de los casos de PAFA, se encuentran implicadas al gen MUTYH, entidad de herencia autosómica recesiva, debido a la presencia de mutaciones germinales bialélicas del cromosoma 1.3,18,19 Esto explica la presencia de antecedentes familiares en la totalidad de los pacientes estudiados. Half y otros19 precisan que aproximadamente el 70 % de los pacientes con PAF, tienen antecedentes familiares de poliposis adenomatosa y de CCR.
Según Itzkowitz y otros,1 la manifestación clínica más frecuente en los pacientes con pólipos, es el sangrado rectal -como aparece en nuestra serie- seguido por la diarrea crónica y el dolor abdominal, este último posiblemente en relación con invaginaciones intestinales a repetición. Moreira y otros3 y Half y otros19 también precisan que en pacientes sintomáticos la rectorragia constituye una de las manifestaciones más frecuentes. Así mismo, Erdman y otros7 coinciden en que el sangrado rectal, las diarreas crónicas y el dolor abdominal son los síntomas más frecuentes de esta afección. En esta enfermedad están descritas 2 formas de presentación, la forma florida (más frecuente), y la forma atenuada, que se caracteriza por presentar menos cantidad de pólipos,1,20 lo cual coincide con nuestro estudio.
Alfaro y otros16 precisan en su estudio de 243 pacientes, que el 52,3 % correspondía a la forma clásica y 47,7 % a la atenuada. Se han realizado estudios que intentan correlacionar las formas de aparición florida o atenuada, con el tipo de mutación presente en cada familia. Bisgaard y otros21 opinan que los pacientes con mutaciones en uno de los dominios de APC, tendrán, a edad muy temprana, el inicio de aparición de adenomas (8,4 años de edad), y que el riesgo de encontrar más de 1 000 adenomas colorrectales fue significativamente alto (odds ratio 3,28).
La displasia exhibida en los adenomas colorrectales recientemente se clasifica en solo 2 grados: displasia de bajo grado -que incluye la displasia leve y moderada- y la displasia de alto grado -que abarca la displasia severa y el carcinoma in situ-.1 Alfaro y otros16 identificaron en el 28 % adenomas colorrectales con displasia de alto grado, resultado que coincide con los encontrados en nuestro estudio.
Los pacientes con PAF nacen con una mutación del APC en la línea germinal en todas las células del cuerpo, así que los tumores se suelen desarrollar en otros órganos además de en el colon. Alfaro y otros16 identificaron lesiones gástricas en el 18,1 % de sus pacientes, y afectación duodenal en el 18,9 %, resultados que se acercan a los que encontramos, a pesar de las diferencia en el número de pacientes estudiados.
Itzkowitz y otros1 y Vasen y otros14 describen que en 30 a 100 % de los pacientes con PAF aparecen pólipos a nivel del estómago, lo cual coincide con nuestra serie. La mayor parte de los pólipos gástricos son no neoplásicos de glándulas fúndicas, y son en los casos típicos, sésiles que miden entre 1 y 5 mm, y al microscopio se caracterizan por hiperplasia de las glándulas fúndicas y microquistes. Pueden aparecer durante la primera época de la vida, incluso, antes de que se desarrollen otros adenomas gastrointestinales. Estas lesiones rara vez muestran evidencia de displasia epitelial, y tienen escaso potencial maligno, como también pueden aparecer microcarcinoides en el estómago. Jiménez Rodríguez y otros22 describen que los adenomas gástricos son infrecuentes, y aparecen en alrededor del 5 % de los pacientes con PAF, por lo general en el antro gástrico. Los adenomas duodenales aparecen en 60 a 90 % de los pacientes con la PAF, y la incidencia aumenta con la edad, con una tendencia de que los adenomas comprometan la región periampular, e incluso, llegar a obstruir el sistema de drenaje pancreático o biliar y provocar pancreatitis.
En un estudio realizado en 14 pacientes con PAF por Katsinelos y otros23 precisaron en el 64,3 % pólipos adenomatosos en el duodeno y en el 50 y 57,1 % en el yeyuno e íleon respectivamente. Vasen y otros,14 Douma y otros24 y Jiménez Rodríguez y otros,22 refieren que si los adenomas duodenales no son tratados precozmente progresaran a la malignidad en el 5 % de los casos. La discrepancia con nuestros hallazgos pudiera justificarse porque nuestros pacientes son de edad pediátrica.
Moreira y otros,3 Andreu y otros5 y Vasen y otros,14 reportan que la HCERP es la manifestación extraintestinal más frecuente, seguida por los dientes supernumerarios y los quistes dentígenos, lo cual coincide con nuestros resultados. Según Pang y otros,25 la HCERP existe casi exclusivamente en la PAF, y se ha relacionado con mutaciones entre el codón 413 en el exón 9, y el codón 1387 en el exón 15. Valanzano y otros26 refieren que los factores genéticos influyen en la expresión clínica de la HCERP, pero no es el único factor responsable del grado de lesión ocular que presentan estos pacientes.
En un estudio cubano realizado por Jiménez Mesa y otros,27 en 14 pacientes sobre el valor de la ortopantomografía en la poliposis familiar de colon, se encontró la presencia de osteomas mandibulares en el 78 %. En el nuestro este hallazgo radiológico no fue precisado, sin embargo se encontró la presencia de quistes dentígenos y dientes supernumerarios, también descritos en esta entidad.
Según Sxott y otros28 y Vasen y otros14 los tumores desmoides son raros, y se presentan solo en 10 a 15 % de los pacientes con PAF. Moraes Righetti y otros15 plantean que es la segunda causa más común de muerte en estos pacientes; nosotros, en nuestra serie, no los encontramos, lo que puede estar relacionado con el número de casos estudiados y la edad temprana de los pacientes. Alfaro y otros16 identificaron en 9,9 % de los pacientes tumores desmoides, en 3,3 % cáncer de tiroides, en 2,4 % osteomas y en 0,4 % tumor cerebral. Groen y otros29 y Celta y otros30 plantean que es imprescindible prestar mayor atención a la aparición de las manifestaciones extraintestinales, que pueden dar al traste con la vida de estos enfermos.
Douma y otros24 refieren que una colectomía preventiva es recomendada cuando los adenomas llegan a ser muy numerosos o grandes, ya que reflejan un riesgo aumentado de CCR, y que el tratamiento quirúrgico es frecuentemente indicado entre las edades de 15 a 25 años. Saab y otros8 informan del desarrollo de CCR en un paciente con PAF en edad tan temprana como los 5 años, y en varios casos en la edad pediátrica, lo que nos obliga al seguimiento estricto de estos pacientes, aun en las primeras décadas de la vida, como se expresa en el trabajo, en el que la indicación de la colectomía estuvo basada en la aparición de displasia de alto grado.
La mayor frecuencia en la realización de la anastomosis ileorectal en el estudio se debe a que es la técnica que más se ha practicado en nuestro medio, al margen de que en estos pacientes se mantiene un 5 % de posibilidades de aparición de CCR en el recto, lo que obliga a su revisión sistemática. La anastomosis ileoanal es la técnica de elección en estos pacientes, pero desde el punto de vista práctico constituye una operación compleja.1 Groen y otros29 y Celta y otros30 relacionan la mayor longevidad de estos pacientes, con la existencia de estrategias de seguimiento y tratamiento.
En nuestra serie, la mayoría de los pacientes estudiados tenían antecedentes familiares de PAF, fueron de color de la piel blanca, las manifestaciones clínicas comenzaron en la segunda década de la vida, y la pesquisa familiar y el sangrado rectal fueron los principales motivos de estudio. Todos los niños presentaron la forma florida de la enfermedad, y en su gran mayoría, displasia de bajo grado en el momento del diagnóstico. Los pólipos extracolónicos se presentaron con mayor frecuencia en el estómago, fueron todos hiperplásicos, y la manifestación extraintestinal más frecuente fue la hipertrofia congénita del epitelio retiniano. La mayoría de los pacientes no se habían realizado colectomía en el momento del estudio. Este trabajo profundiza en la necesidad del tratamiento integral de esta enfermedad, así como su seguimiento, para lograr un mejor diagnóstico, tratamiento y supervivencia en estos pacientes.
REFERENCIAS BIBLIOGRÁFICAS
1. Itzkowitz S, Potack J. Colonic Polyps and Polyposis Syndromes. In: Feldman M, Friedman L, Brandt L. Sleisenger and Fordtran's Gastrointestinal and Liver Disease. 9th ed. Philadelphia: Editorial Saunders Elsevier; 2010. p. 2176-87.
2. Shilyansky J. Tumores del tracto digestivo. En: Behrman RE, Kliegman RM, Jenson HB. Nelson. Tratado de Pediatría. 17a. ed. Philadelphia: Editorial Elsevier; 2004. p. 1290-1.
3. Moreira L, Castells A, Castelví S. Pólipos y poliposis colorrectales. En: Montoro MA, García Pagán JC. Gastroenterología y Hepatología. Problemas Comunes en la Práctica Clínica. 2da. ed. Barcelona: Jarpyo Editores S.A.; 2012. p. 607-16.
4. Arévalo F, Aragón V, Alva J, Pérez M, Cerrillo G, Montes P. Pólipos Colorectales: actualización en el diagnóstico [revisión]. Rev Gastroenterol Perú. 2012;32-2:123-33.
5. Andreu M, Ferrández A. Pólipos colorrectales y poliposis intestinal. En: Ponce J, ed. Tratamiento de las enfermedades gastroenterológicas. 3era. ed. Barcelona: Doyma; 2011. p. 345-58.
6. Graziano A, Gutiérrez A. Poliposis Adenomatosa Familiar. En: Reis Neto JA. Coloproctología Atual. Proctosite Colorectal Surgery At the Web [libro en Internet] Capítulo III; 2008 [citado 2 de enero de 2012]. Disponible en: http://www.proctosite.com/library/books/livro_reis_novo/cap03.pdf
7. Erdman SH, Hoffenberg EJ. Polyps and Tumors of the Gastrointestinal Tract. In: Rudolph CD, Rudolph A, Lister GE, First L, Gershon A. Rudolph's Pediatrics Section 21. Disorders of the Gastrointestinal System. Parte 5. Disorders of the Stomach and Intestine. 22nd. ed. New York: McGraw-Hill; 2011. p. 414.
8. Saab R, Furman WL. Epidemiology and Management Options for Colorectal Cancer in Children. Pediatr Drugs. 2008;10(3):177-92.
9. McCart A, Latchford A, Volikos E, Rowan A, Tomlinson I, Silver A. A novel exon duplication event leading to a truncating germ-line mutation of the APC gene in a familial adenomatous polyposis family. Fam Cancer. 2006;5(2):205-8.
10. Bisgaard ML, Bülow S. Familial adenomatous polyposis (FAP): genotype correlation to FAP phenotype with osteomas and sebaceous cysts. Am J Med Genet A. 2006 Feb 1;140(3):200-4.
11. Hegde MR, Roa BB. Detecting mutations in the APC gene in familial adenomatous polyposis (FAP). Curr Protoc Hum Genet. 2006 Aug;10:108.
12. Kanter-Smoler G, Fritzell K, Rohlin A, Engwall Y, Hallberg B, Bergman A, et al. Clinical characterization and the mutation spectrum in Swedish adenomatous polyposis families. BMC Med. 2008 Apr 24;6:10.
13. Stekrova J, Sulova M, Kebrdlova V, Zidkova K, Kotlas J, Ilencikova D, et al. Novel APC mutations in Czech and Slovak FAP families: clinical and genetic aspects. BMC Med Genet. 2007 Apr 5;8:16.
14. Vasen H, Möslein G, Alonso A, Aretz S, Bernstein I, Bertario L, et al. Guidelines for the clinical management of familial adenomatous polyposis (FAP). Gut. 2008;57:704-13.
15. Moraes Righetti AE, Jacomini C, Serafim Parra R, Ribeiro de Almeida Normanha AL, Ribeiro Rocha JJ. La poliposis adenomatosa familiar y tumores desmoides. Clínicas (Sao Paulo). 2011 Octubre;66(10):1839-42.
16. Alfaro I, Ocaña T, Castells A, Cordero C, Ponce M, Cajal RY, et al. Características de los pacientes con poliposis adenomatosa familiar en España. Los primeros resultados del Registro Español de Poliposis Adenomatosa Familiar. Med Clin (Barc). 2010 Jun 19;135(3):103-8.
17. Björk J. Epidemiología de la poliposis adenomatosa familiar en Suecia: evolución en el tiempo y las diferencias en el fenotipo entre varones y mujer. El Registro de Poliposis Sueco. Departamento de Gastroenterología y Hepatología. Hospital Karolinska Suecia. 1999;34(12):1230-5.
18. Grau García C, Soto Gutiérrez A, Andrada Becerra E, Sánchez de las Heras B, Gallego Plazas J, Brotons Brotons A, et al. Paciente con poliposis adenomatosa familiar y metástasis hepáticas de tumor neuroendocrino. Gastroenterología y Hepatología. 2011;34(5):329-32.
19. Half E, Bercovich D, Rozen P. Familial adenomatous polyposis. Orphanet Journal of Rare Diseases. 2009;4:22.
20. Stella A, Resta N, Gentile M, Susca F, Mareni C, Montera MP, et al. Exclusion of the APC gene as the cause of a variant form of familial adenomatous polyposis (FAP). Am J Hum Genet. 1993 Nov;53(5):1031-7.
21. Bisgaard ML, Ripa R, Knudsen AL, Bülow S. Familial adenomatous polyposis patients without an identified APC germline mutation have a severe phenotype. Gut. 2004 Feb;53(2):266-70.
22. Jiménez Rodríguez RM, Suárez Artacho G, Morcillo J, Díaz Pavón JM, Morales Méndez S. Adenocarcinoma de cuarta porción de duodeno en paciente con poliposis adenomatosa familiar. Rev Esp Enferm Dig. 2007 Ago;99(8):477-8.
23. Katsinelos P, Kountouras J, Chatzimavroudis G, Zavos C, Pilpilidis I, Fasoulas K, et al. Wireless capsule endoscopy in detecting small-intestinal polyps in familial adenomatous polyposis. World Journal Gastroenterology. 2009;15(48):6075-9.
24. Douma KFL, Bleiker EMA, Aaronson NK, Cats A, Gerritsma MA, Gundy CM. Long-term compliance with endoscopic surveillance for familial adenomatous polyposis. Colorectal Disease. 2010;12:1198-207.
25. Pang CP, Keung JW, Tang NL, Fan DS, Lau JW, Lam DS. Congenital hypertrophy of the retinal pigment epithelium and APC mutations in two Chinese families with familial adenomatous polyposis. Eye (Lond). 2000 Feb;14(Pt 1):18-22.
26. Valanzano R, Cama A, Volpe R, Curia MC, Mencucci R, Palmirotta R, et al. Congenital hypertrophy of the retinal pigment epithelium in familial adenomatous polyposis. Novel criteria of assessment and correlations with constitutional adenomatous polyposis coli gene mutations. Cancer. 1996 Dec 1;78(11):2400-10.
27. Jiménez Mesa G, López Barbán MA, Hano García O. Valor de la ortopantomografía en la poliposis familiar de colon. Rev Cubana Med. 1996 Abr;35(1):24-9.
28. Sxott RJ, Froggatt NJ, Trembath RC, Evans Evans DG, Hodgson SV, Maher ER. Familial infiltrative fibromatosis (desmoid tumours) (MI M135290) caused by a recurrent 3' APC gene mutation. Hum Mol Genet. 1996 Dec;5(12):1921-4.
29. Groen EJ, Roos A, Muntinghe FL, Enting RH, de Vries J, Kleibeuker JH, et al. Extra-intestinal manifestations of familial adenomatous polyposis. Ann Surg Oncol. 2008 Sep;15(9):2439-50.
30. Celta F, Dhamo A, Civitelli S, Zangari R. Comment on Extraintestinal manifestations of familial adenomatous polyposis. Ann Surg Oncol. 2009;16(5):1446-8.
Recibido: 10 de enero de 2014.
Aprobado: 18 de febrero de 2014.
Tulio Antonio Amaya Sorto. Instituto de Gastroenterología. Calle 25, entre H e I, Vedado, municipio Plaza. La Habana, Cuba. Correo electrónico: tulioamayasorto@hotmail.com

{kind=link}