










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Pediatría
versión impresa ISSN 0034-7531
Rev Cubana Pediatr vol.86 no.3 Ciudad de la Habana jul.-set. 2014
PRESENTACIÓN DE CASOS
Hiperplasia adrenal congénita en forma clásica virilizante simple
Congenital adrenal hyperplasia in simple classical virilizing form
MSc. María del Carmen Valdés Alonso,I Dr. José María Basain Valdés,II Dra. Yadenis Bioti TorresI
IHospital Pediátrico Docente "Juan Manuel Márquez". La Habana, Cuba.
IIInstituto Nacional de Endocrinología. La Habana, Cuba.
RESEUMEN
El déficit de 21-hidroxilasa es la forma más frecuente de hiperplasia adrenal congénita, que forma parte de los desórdenes de la diferenciación sexual. Se presentan 3 casos. El primero, un recién nacido de 19 días que es llevado a consulta por presentar desórdenes de los genitales externos. Al examen físico presentaba un clítoris aumentado de tamaño, con orificio uretral en su base y engrosamiento de los rodetes labioescrotales. El diagnóstico se realizó por ultrasonido ginecológico, cromatina sexual, estudios hormonales y cariotipo. El segundo caso, un recién nacido de 15 días que también es llevado a consulta por desórdenes de los genitales externos, con examen físico similar al primer caso, y se le realizaron los mismos complementarios para su diagnóstico. El tercer caso, un lactante de 2 meses de edad, que es llevado a consulta por igual motivo, y que al examen físico se encontró hiperplasia del clítoris, con orificio en su base, y engrosamiento de los labios mayores que estaban fusionados en la línea media. Se le indicaron iguales complementarios. Se diagnosticó en los 3 casos una hiperplasia adrenal virilizante, y se realizó tratamiento sustitutivo hormonal y cirugía reconstructiva de los genitales externos.
Palabras clave: hiperplasia adrenal congénita, déficit de 21-hidroxilasa, desórdenes de la diferenciación sexual, 17-OH progesterona.
ABSTRACT
Steroid 21-hydroxylase is the most frequent form of congenital adrenal hyperplasia that is part of the sexual differentiation disorders. This article reported 3 cases. The first one was a 19 days-old infant who was taken to the doctor´s because of external genitalia disorders. The physical exam revealed augmented clitoris with urethral orifice in its basis and thickening of the labioscrotal swellings. The patient was diagnosed by means of gynecological ultrasound, sexual chromatin, hormonal studies and karyotype. The second case was a 15 days-old newborn, who was also taken to the doctor´s for external genitalia disorders. The physical exam was similar to that of the first case and the same complementary tests were performed for diagnosis. The third case was a 2 months-old infant who was taken to the medical service for the same reasons, and his physical exam showed clitoris hyperplasia, orifice in its base and thickening of labia majora that fused in the midline. The same complementary tests were indicated. The final diagnosis in these three cases was virilizing adrenal hyperplasia. They were all treated with hormone replacement therapy and reconstructive surgery of their external genitalia.
Keywords: congenital adrenal hyperplasia, steroid 21-hydroxylase, sexual differentiation disorders, 17 OH progesterone.
INTRODUCCIÓN
El déficit de 21-hidroxilasa representa del 90 al 92 % de todas las causas de hiperplasia adrenal congénita. La incidencia de la enfermedad es, en términos medios, de 1:5 000 nacimientos, con marcadas variaciones étnicas.1 Es una enfermedad autosómica recesiva, en la cual, según Dupont y otros, existe una estrecha relación genética entre el gen de la 21 hidroxilasa y el locus B del sistema mayor de histocompatibilidad y la expresión fenotípica.2,3 Las formas clínicas del déficit en 21-hidroxilasa están asociadas con una afección del gen que codifica el citocromo P450c21, que fue descubierto en 1984, y se localizó en el brazo corto del cromosoma 6. En realidad, existen 2 genes (el CYP21A y el CYP21B), y la gran frecuencia de recombinaciones e intercambios entre los genes se correlaciona con el grado de afectación de la actividad de la 21-hidroxilasa, así como con la forma de presentación de la enfermedad.4
Se clasifican en 2 grandes grupos: intraútero (formas clásicas) y etapa posnatal (formas no clásicas), y se considera un tercer grupo que incluye las formas crípticas. Dentro de las formas clásicas se incluye la variedad perdedora de sal y la virilizante simple; mientras, en las formas no clásicas, se consideran, en la infancia, la seudopubertad precoz, y en la pubertad, las presentaciones de hirsutismo, acné, trastornos menstruales, así como la infertilidad en la adolescencia tardía y adultez. Las formas crípticas son asintomáticas.4
Desde el punto de vista clínico las formas clásicas (congénitas) por definición, son las formas severas de la enfermedad que se expresan ya en el útero. En el periodo crítico de diferenciación de los órganos genitales externos, el cojinete genital es muy sensible a la acción de este exceso de testosterona y dihidrotestosterona, y por ello, los fetos femeninos se virilizan. Por el contrario, en el feto masculino, que tiene una secreción testicular normal, un aporte suplementario de testosterona/dihidrotestosterona carece de efecto sobre su diferenciación sexual, pero sí influye en el tamaño del pene por acción de los andrógenos.5-7
A continuación se presentan 3 casos, cuyo interés radica no solo en la magnitud de la afección, sino en su forma de presentación.
PRESENTACIÓN DE CASOS
Caso 1
Recién nacido de 19 días, que es llevado a consulta por presentar desórdenes en la diferenciación de los genitales externos. En el interrogatorio se recoge que es hijo de padres no consanguíneos, nacido por parto eutócico a las 38 semanas de edad gestacional, con peso de 3 230 g, talla de 50 cm, circunferencia cefálica de 34 cm y Apgar 9/9. La madre tiene 25 años, y no tiene antecedentes patológicos personales ni familiares a destacar (gestación 1, paridad 1, aborto 0).
El seguimiento del embarazo se realizó según lo establecido por el Programa Materno Infantil, con ecografías normales, no se recoge el antecedente de ingerir medicamentos, ni de exponerse a agentes teratogénicos. La paciente procedía de un área rural, y se le realizó la pesquisa neonatal, pero la familia decide -ante este problema- acudir a nuestro centro y no nos aportan los valores de la 17-hidroxiprogesterona de la pesquisa. El examen físico de los genitales externos de la recién nacida reflejó un tubérculo genital aumentado de tamaño, con orificio uretral en su base, engrosamiento de los rodetes labioescrotales, y no se palparon las gónadas en dichas estructuras ni en el canal inguinal.
Se le indicó una ecografía ginecológica, en la que se observó el útero con características normales para la edad. La cromatina sexual arrojó 20 % de cuerpos de Barr. El cariotipo reportó 46, XX en 12 metafases. El nivel de 17 hidroxiprogesterona al duodécimo tercer día de vida fue de 84 ng/mL. La tasa basal de testosterona fue de 6 nmol/mL. Se consideró que este paciente era del sexo femenino, y se concluyó el diagnóstico de hiperplasia adrenal congénita en forma clásica virilizante simple. Se inició tratamiento con cortisona a razón de 20 mg/m2/día, y fue evaluado por el equipo multidisciplinario constituido por las especialidades de Cirugía, Urología, Neonatología, Endocrinología y Genética. Se programó el seguimiento por consulta y la cirugía correctora de los genitales externos, que se realizó después de los 12 meses de edad, y se dejó pendiente la vaginoplastia para la etapa puberal.
Caso 2
Paciente de 15 días de nacido, que es llevado a consulta por presentar alteraciones del desarrollo de los genitales externos. Es hijo de padres no consanguíneos, nacido por parto eutócico, a las 40 semanas de edad gestacional, con un peso de 2 980 g, talla de 49 cm, circunferencia cefálica de 36 cm y Apgar 9/10. La madre es joven, y no se recogen antecedentes de padecer ninguna enfermedad, pero sí existen antecedentes familiares de hipertensión arterial (abuela paterna). La historia obstétrica recoge 2 embarazos y ningún aborto.
Según lo establecido por el Programa Materno Infantil se realizó el seguimiento normal del embarazo. No hubo ingestión de fármacos por la madre que causaran virilización al feto, así como no se expuso a agentes ambientales teratógenos, y los estudios imagenológicos fueron normales. La paciente procedía de la provincia del interior del país.
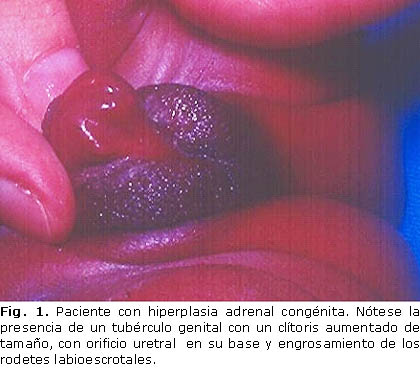
El examen físico de los genitales externos de la recién nacida reflejó un tubérculo genital con un clítoris aumentado de tamaño, con orificio uretral en su base, engrosamiento de los rodetes labioescrotales (Fig. 1), y no se palparon las gónadas en estos, ni en la región inguinal.
Se le indicó una ecografía ginecológica, en la que se observó el útero con características normales para la edad. La cromatina sexual arrojó 18 % de cuerpos de Barr. El cariotipo reportó 46, XX en 12 metafases. El nivel de 17-hidroxiprogesterona al noveno día de vida fue de 90 ng/mL. La tasa basal de testosterona fue de 7 nmol/mL, por lo que se diagnosticó hiperplasia suprarrenal congénita en forma clásica virilizante simple, y se inició tratamiento con cortisona a razón de 20 mg/m2/día. Posteriormente se discutió en colectivo por el equipo multidisciplinario (Cirugía, Urología, Neonatología, Endocrinología y Genética). Se programó su seguimiento por las consultas, y la cirugía correctora de los genitales que se realizó después de los 12 meses de edad. Quedó pendiente la reconstrucción de vagina en la etapa puberal.
Caso 3
Lactante de 2 meses de edad, que es llevado a consulta por presentar alteración de los genitales externos. Nace producto de parto eutócico, a las 39 semanas de edad gestacional, con peso de 2 889 g, talla de 52 cm, circunferencia cefálica de 36 cm y Apgar 9/9. La madre tiene 30 años, no refiere padecer de ninguna enfermedad antes o durante el embarazo, así como llevar algún tratamiento hormonal para lograr la gestación, como tampoco haberse expuesto a agentes teratógenos. Existen antecedentes patológicos familiares de obesidad y dislipidemia (abuelo paterno) y no de consanguinidad con el padre; tuvo 3 embarazos, y uno de ellos fue aborto provocado.
Refiere haber asistido a todas las consultas programadas durante el embarazo, y se realizó todos los complementarios indicados según se establece en el Programa Materno Infantil. La paciente procedía de área rural de una provincia del interior del país.
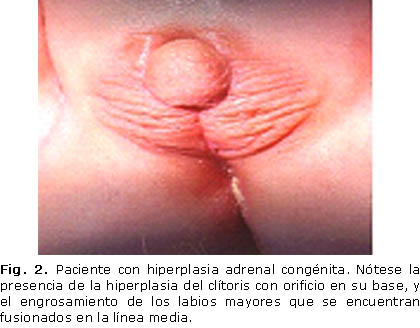
Al examen físico de genitales externos del recién nacido, se reflejó hiperplasia del clítoris, con orificio en su base, engrosamiento de los labios mayores que estaban fusionados en la línea media (Fig. 2), y las gónadas no se palparon en esos labios fusionados ni en el canal inguinal.
Se indicó una ecografía ginecológica, en la que se observó el útero con características normales para la edad. La cromatina sexual arrojó 21 % de cuerpos de Barr. El cariotipo reportó 46, XX en 12 metafases. El nivel de 17-hidroxiprogesterona a los 2 meses de vida fue de 99 ng/mL, la tasa basal de testosterona fue de 9,5 nmol/mL. Se consideró que se trató de un recién nacido del sexo femenino, se concluyó con el diagnóstico hiperplasia adrenal congénita en forma clásica virilizante simple, y se inició tratamiento con cortisona a razón de 20 mg/m2/día. Se interconsultó con las especialidades de Cirugía, Urología, Endocrinología y Genética. Se programó el seguimiento por consulta, y posteriormente se realizó la cirugía correctora de los genitales externos después de los 12 meses de edad. Quedó pendiente la corrección quirúrgica de la vagina para la etapa puberal.
DISCUSIÓN
En nuestros 3 casos se trató de una forma clásica de hiperplasia adrenal congénita por déficit de 21-hidroxilasa, lo que implica la existencia de un hiperandrogenismo ya intraútero que condiciona la aparición de genitales grandes en el varón, y de un grado variable de virilización de los genitales externos en la niña.8
En la forma clásica variedad perdedora de sal (que es la expresión más grave de la enfermedad) existe un déficit de cortisol y de aldosterona, y se manifiesta en ambos sexos con crisis de pérdida salina en la época neonatal, con cuadros de deshidratación, vómitos, fallo de medro, etc. Esta crisis de insuficiencia suprarrenal tiene una importante morbilidad y mortalidad si no se instaura un tratamiento adecuado, lo cual no ocurrió en estos pacientes, pues se trataba de una forma clásica virilizante, pero siempre debe realizarse un examen minucioso de los genitales externos de los pacientes, y brindar a la familia -por el neonatólogo o por el pediatra del equipo de salud- toda la información necesaria.8
En la forma clásica virilizante simple, la síntesis de aldosterona no está tan gravemente alterada, por lo que se mantiene la homeostasis del sodio y no presentan crisis de pérdida salina. Las niñas son identificadas precozmente por la virilización de los genitales externos, pero los niños, y las niñas -con una virilización leve- suelen diagnosticarse tardíamente en la infancia, cuando ya se hacen manifiestos los signos de hiperandrogenismo y la aparición de una seudopubertad precoz. En estos casos se trató de una forma clásica virilizante, que se diagnosticó en las niñas por los signos de hiperandrogenismo existentes en los pacientes.9
Algunas formas no clásicas presentan un hiperandrogenismo de aparición posnatal. Los síntomas más frecuentes en la infancia son pubarquia prematura, piel grasa con acné, aceleración del crecimiento y de la maduración ósea; en las niñas, puede aparecer una moderada hipertrofia del clítoris. En la adolescencia y la edad adulta las mujeres pueden presentar irregularidades menstruales, hirsutismo, calvicie, ovario poliquístico, acné e infertilidad. Los varones afectados pueden presentar acné, oligospermia e infertilidad, pero la mayoría de las veces son asintomáticos, lo cual no se correspondió con los casos descritos.10
Las formas crípticas cursan únicamente con hallazgos hormonales, pero sin ninguna sintomatología, si bien actualmente se cree que pueden presentar, eventualmente, algún signo clínico de hiperandrogenismo.10
El déficit de 3β-hidroxiesteroide deshidrogenasa puede cursar, de manera similar, al déficit de 21-hidroxilasa con pérdida salina y/o virilización de los genitales externos con genitales internos normales, pero, en estos pacientes, los niveles de 17-hidroxiprogesterona son normales, y el diagnóstico se realizaría por la determinación de dehidroepiandosterona (DHEA), lo que resulta poco frecuente.11
Con respecto al déficit de 11β-hidroxilasa es raro, y aunque cursa con virilización de los genitales externos -y en ocasiones con hipercaliemia- existe siempre hipertensión arterial asociada, ya que, dado el déficit de la enzima existe un aumento del metabolito por encima del déficit (desoxicortisol), lo que causa el incremento de la tensión arterial en estos pacientes, que no se manifestó en nuestros casos.7,11
La deficiencia de 20-hidroxilasa es una condición muy rara de ocurrencia, compromete el paso de colesterol a pregnenolona, y la síntesis de los 3 tipos de esteroides suprarrenales, lo que lleva a un acúmulo de grandes cantidades de colesterol y ésteres de colesterol, que se ve en la glándula como células corticales repletas de material lipoide, por lo que se denomina hiperplasia adrenal lipoide. Hay ausencia de genitales externos masculinos; las mujeres afectadas tienen desarrollo genital normal, la falta de mineralocorticoides conduce a pérdida severa de sodio, hipovolemia e hipotensión. La mayoría de los pacientes no sobrepasa la infancia temprana.11
En la deficiencia de 18-hidroxiesteroide deshidrogenada solo se compromete la producción de aldosterona, lo que produce pérdida de sodio hiponatremia, hiperkalemia y deshidratación, y no presentan virilización o mal diferenciación sexual de los genitales externos.11
Por último, hay que tener presente el hermafroditismo verdadero en su variedad 46, XX que en la ecografía ginecológica arroja útero de aspecto normal acorde con la edad de la paciente, pero no tiene niveles elevados de 17-hidroxiprogesterona, y generalmente se encuentra una gónada en el lado contrario al ovario o en región inguinal.11
Todos los pacientes con déficit clásico de 21-hidroxilasa, así como los pacientes sintomáticos de las formas no clásicas, deben tratarse con glucocorticoides, ya que así se suprime el exceso de secreción de hormona liberadora de corticotropina y se reduce el exceso de esteroides sexuales de origen adrenal.12
La hidrocortisona es el tratamiento más fisiológico (tiene una potencia similar a la del cortisol endógeno, una corta vida biológica que minimiza el efecto sobre el crecimiento y sobre otros efectos adversos). La dosis es de 10 mg/m2/día, variable en función de la edad. Los neonatos son tratados habitualmente a una dosis de 20 mg/m2/día, dividido en 3 subdosis en las formas clásicas de presentación de la enfermedad. En los pacientes con formas no clásicas sintomáticas, está indicado iniciar el tratamiento con glucocorticoides a dosis bajas, generalmente a mitad de dosis que en las formas clásicas.3
Los pacientes con pérdida salina requieren la administración de un mineralocorticoides, el más utilizado es el 9 alfa fluorhidrocortisona, 0,1 mg/día, y en ocasiones se requieren suplementos de cloruro de sodio (1-2 g/d), durante el primer año de vida.12 En situaciones de estrés o de enfermedad intercurrente, los pacientes requieren tratamiento hidroelectrolítico apropiado y aumentar la dosis de hidrocortisona, que debe administrarse por vía intravenosa o intramuscular.8
Respecto al tratamiento quirúrgico, la actitud terapéutica se inicia con la asignación precoz del sexo que debe ser la del sexo genético/gonadal, por la posibilidad de mantener la función reproductora. Se obtienen buenos resultados con la realización de la reconstrucción genital (clitoroplastia y vaginoplastia) en un mismo acto quirúrgico, hacia el segundo semestre de edad, según plantean algunos autores, aunque otros consideran realizar la vaginoplastia en la pubertad. El objetivo es la corrección de los genitales externos antes de los 18 meses de edad.13
El buen control terapéutico durante la infancia y la adolescencia es fundamental para asegurar un crecimiento correcto, una maduración puberal normal y una ausencia de complicaciones a largo plazo. El objetivo es buscar la dosis mínima eficaz que garantice un buen crecimiento y una adecuada supresión de los andrógenos suprarrenales. Los parámetros de vigilancia incluyen datos clínicos como: edad ósea, peso, talla y velocidad de crecimiento y hormonales, como determinación de 17-hidroxiprogesterona, testosterona, delta 4 androstenediona, ACTH y actividad de renina plasmática.10
En las gestaciones con riesgo de tener un hijo afectado por hiperplasia suprarrenal virilizante se ha conseguido frenar la producción de andrógenos suprarrenales fetales, y disminuir la ambigüedad genital administrando dexametasona a la madre gestante. De esta manera se previene la virilización genital del feto mujer afectado.12 El tratamiento prenatal debe ir acompañado siempre de un adecuado diagnóstico genético prenatal.14
Desde el año 2000 en nuestro país se inició la pesquisa de la hiperplasia adrenal congénita por déficit de 21-hidroxilasa, mediante el test de determinación de 17-OH progesterona en sangre de talón.15
En los 2 primeros casos estudiados, los familiares no aportaron los valores de la pesquisa, pues acudieron al centro de forma rápida en busca de opciones terapéuticas. En el tercer caso la mamá refiere que se realizó la pesquisa, pero acudió a nuestro centro en busca de más información y de un tratamiento para su niña, sin precisar los valores obtenidos durante la pesquisa neonatal. Todos estos pacientes procedían de área rural de municipios que no pertenecían a La Habana.
Los autores comparten la idea de que el neonatólogo y el pediatra deben examinar los genitales externos de los recién nacidos, con el objetivo de diagnosticar, precozmente, la enfermedad, y poder brindar a los familiares la información adecuada, así como explicar que esta será tratada por un equipo multidisciplinario para una correcta asignación del sexo y garantizar una mejor calidad de vida de los pacientes. Se trata, en todo momento, de evitar la ansiedad familiar.
REFERENCIAS BIBLIOGRÁFICAS
1. Pérez Samper LA, Martínez Ramos NR. Hiperplasia suprarrenal congénita por déficit de la 21 hidroxilasa. Presentación de un caso. MEDICIEGO. 2013;19(Supl. 1):124-34.
2. Bose HS, Sugawara T, Strauss JF. The pathophysiology and genetics of congenital lipoid adrenal hyperplasia. N Engl J Med. 2006;335:1870-8.
3. Minutolo C, Nadra AD, Fernandez C, Taboas M. Structure-Based Analysis of Five Novel Disease-Causing Mutations in 21-Hydroxylase-Deficient Patients. PLoS One. 2011;6(1):158-99.
4. Chancia S, Fulidenberg G, Jinesk L. Ocurrence of male phenotype in genotypic with congenital virilizing adrenal hyperplasia. Am J Med Genet. 2009;34:406-12.
5. Marino R, Ramirez P, Galeano G, Perez Garrido N, Rocco C, Ciaccio M, et al. Steroid 21-hydroxylase gene mutational spectrum in 454 argentinean patients: genotype-phenotype correlations in a large cohort of patients with congenital adrenal hyperplasia. Clin Endocrinol. 2011;75:427-35.
6. Krone N, Arlt W. Genetics of congenital adrenal hyperplasia. Best Pract Res Clin Endocrinol Metab. 2009;23:181-92.
7. Miller WL. Pathophysiology, genetics, and treatment of hyperandrogenism. Pediatr Clin North Am. 1997;44:375-95.
8. Acevedo J, Acevedo S. Hiperplasia Suprarrenal Congénita Virilizante. Rev Ped Elec. 2012;9(2):193-8.
9. Speiser P, Azziz R, Baskin L, Ghizzoni L, Hensle T, Merke D. Congenital Adrenal Hyperplasia due to steroid 21-hydroxylase deficiency: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2010;95:4133-60.
10. Pasqualini T, Alonso G, Fernández C, Buzzalino N, Dain L. Talla final en varones sintomáticos con hiperplasia suprarrenal no clásica tratados con glucocorticoides. Casos clínicos. Arch Argent Pediatr. 2013;111(2):86-94.
11. Henao G. Hiperplasia Suprarrenal. En: Botero J, Jubiz A, Henao G. Obstetricia y Ginecología. 5ta. ed. Medellín, Colombia: Editorial Carvajal S.A.; 2007. p. 414-9.
12. Mercado AB, Wilson RC, Cheng KC. Extensive personal experience. Prenatal treatment and diagnosis of congenital adrenal hyperplasia owing to steroid 21 hydroxylase deficiency. J Clin Endocrinol Metab. 2008;80:2014-20.
13. Alemán Ramírez R, Céspedes Durán L, Fernández Vaglio R, Herrera Rodríguez A, Sánchez Villalobos N, Solar Del Valle T, et al. Desórdenes del desarrollo sexual y cirugía correctiva. Med Leg Costa Rica. 2013;30(2):134-41.
14. Lajic S, Nordenström A, Hirvikoski T. Long-term outcome of prenatal treatment of congenital adrenal hyperplasia. Endocr Dev. 2008;13:82-98.
15. Coto Rodeiro R, Varona Sánchez JA, Borrego López JA, Formoso Martín LE. Resultados de la pesquisa de hiperplasia adrenal congénita en recién nacidos. Rev Cubana Obstet Ginecol. 2011;37(2):136-46.
Recibido: 28 de noviembre de 2013.
Aprobado: 6 de enero de 2014.
María del Carmen Valdés Alonso. Hospital Pediátrico Universitario "Juan Manuel Márquez". Avenida 31 y calle 76, municipio Marianao. La Habana, Cuba.
Correo electrónico: mavaldes@infomed.sld.cu
