










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Pediatría
versión impresa ISSN 0034-7531
Rev Cubana Pediatr vol.86 no.4 Ciudad de la Habana oct.-dic. 2014
Rev Cubana Pediatr. 2014;86(4)
ARTÍCULO ORIGINAL
Caracterización de la fibrosis quística en el primer año de vida
Characterization of cystic fibrosis in the first year of life
MSc. Gladys Fuentes Fernández, Dr.C. Gladys Abreu Suárez, MSc. Aurelia Paula Pérez Brunet, MSc. José Antonio González Valdés, MSc. Reyna Portuondo Leyva
Hospital Pediátrico Centro Habana. La Habana, Cuba.
RESUMEN
Introducción: la fibrosis quística es una enfermedad multisistémica, genética y letal, con daño pulmonar temprano y progresivo en los casos típicos.
Objetivo: caracterizar el diagnóstico de la fibrosis quística en niños menores de un año.
Métodos: estudio transversal, descriptivo, a pacientes diagnosticados con fibrosis quística según sospecha clínica, en el primer año de vida, en el Hospital Pediátrico Centro Habana, en el período 1993-2013, según datos obtenidos de los registros clínicos.
Resultados: del total de 44 pacientes diagnosticados en el periodo, 35 clasificaron como enfermedad pulmonar típica, de ellos 13 fueron diagnosticados durante el primer año de vida, 7/21 en el periodo 1993-2003 (33,3 %) y 6/14 entre 2004-2013 (42,8 %). Cuando se suman las formas clínicas no típicas, el total de niños diagnosticados durante su primer año fue de 16, con edad media al diagnóstico de 4,9 meses; en 11 niños (68,8 %) el diagnóstico fue antes de los 6 meses, con predominio de los varones (10-62,5 %). La desnutrición estuvo presente en el 62,5 %. La forma típica con insuficiencia pancreática se evidenció en el 75 % de los menores de un año estudiados, y se aisló Pseudomona aeruginosa en el 68,8 %. Predominaron las manifestaciones respiratorias como la polipnea y el tórax hiperinsuflado (15 niños, 93,8 %), así como el tiraje (14 niños, 87,5 %), mientras que la distensión abdominal (11, 68,8 %) y la esteatorrea (10, 62,5 %), fueron las más frecuentes alteraciones digestivas. La radiografía torácica mostró algún grado de alteración en todos los niños.
Conclusiones: es importante una alta sospecha clínica para el diagnóstico temprano de las formas típicas de la fibrosis quística.
Palabras clave: fibrosis quística, pesquisa neonatal, diagnóstico temprano.
ABSTRACT
Introduction: cystic fibrosis is a lethal multystemic and genetic disease causing early progressive pulmonary damage in typical cases.
Objective: to characterize the diagnosis of cystic fibrosis in children under one year of age.
Methods: descriptive cross-sectional study of patients diagnosed with cystic fibrosis in their first year of life, based on clinical suspicions, in the 1993-2013 period at Centro Habana hospital. To this end, data from clinical records were used.
Results: of the total number of 44 patients diagnosed with the disease in the period, 35 were classified as having typical pulmonary disease of whom 13 were diagnosed in their first year of life, 7 of 21 (33.3 %) detected in the 1993-2003 period and 6 of 14 (42.8 %) diagnosed from 2004 through 2013. When the non-typical clinical forms of disease are added, then the total amount of children diagnosed in their first year of life amounts to 16 with average age at diagnosis of 4.9 months; 11 children (68.8 %) were diagnosed before 6 months of age and males prevailed (10 for 62.5 %). Malnutrition was present in 62.5 % of the group. The typical form with pancreatic failure was observed in 75 % of under one year-old children whereas Pseudomona aeruginosa was isolated in 68.8 % of patients. Respiratory symptoms such as polypnea and hyperinsuflated thorax predominated (15 children for 93,8 %) and retraction (14 children for 87.5 %) whereas abdominal distension (11 for 68.8 %) and steatorrhea (10 for 62.5 %) were the most common digestive disturbances. Chest X-ray showed some alteration in all these children.
Conclusions: the medical staff should be highly suspicious of these cases from the clinical viewpoint in order to early diagnose the typical forms of cystic fibrosis.
Keywords: cystic fibrosis, neonatal screening, early diagnosis.
INTRODUCCIÓN
La fibrosis quística (FQ) es una enfermedad multisistémica, que se hereda con carácter autosómico recesivo. Esta afección condiciona un daño pulmonar progresivo desde la etapa de lactante, lo que afecta la calidad de vida y la supervivencia de los individuos afectados.1-3
La supervivencia de los enfermos en el momento actual se ha elevado, gracias a la intervención precoz y agresiva de las complicaciones, el tratamiento nutricional riguroso y el desarrollo de centros con enfoque multidisciplinario, basados en algoritmos de tratamiento.4
La pesquisa neonatal a través de la determinación de tripsina inmunorreactiva, ha posibilitado el diagnóstico en los primeros meses de la vida, con acciones más tempranas dirigidas a favorecer el aclaramiento de las secreciones y garantizar un buen estado nutricional.4-8
Cuba tiene una Comisión Nacional de FQ, que data desde 1974, con centros de atención en todas las provincias del país y 4 en la capital, que son de referencia nacional. Esta Comisión ha dictado pautas para el diagnóstico y tratamiento de los enfermos, y se ocupa, además, de la capacitación sistemática del personal que integran estos grupos multidisciplinarios.2,6
En la actualidad no se ha podido introducir en Cuba el diagnóstico mediante pesquisa neonatal, y es importante, por tanto, elevar la sospecha clínica, de forma de realizar diagnósticos más tempranos, con menos deterioro pulmonar. Esta circunstancia nos ha motivado a evaluar cómo se comporta el diagnóstico en el primer año de la vida en nuestro centro, sobre todo, en niños con formas típicas.
MÉTODOS
Se realizó un estudio descriptivo, transversal, de todos los pacientes diagnosticados con FQ en la consulta del Grupo Multidisciplinario de FQ, en el Hospital Pediátrico Centro Habana, desde el año 1993 hasta 2013, ambos inclusive, sobre la base de sus registros clínicos. Se clasificó a los pacientes en formas típicas o no típicas, según la Clasificación de Dodge Modificada (OMS, 2008),2 y se agruparon según el año de diagnóstico en dos periodos: 1993-2003 y 2004-2013. En los diagnosticados antes del año de edad se estudió el sexo, la edad al diagnóstico en meses, el aislamiento de gérmenes en secreciones obtenidas por hisopado faríngeo profundo, el estado nutricional según los percentiles de peso para la talla de acuerdo con las tablas cubanas de crecimiento y desarrollo, expresados como: delgado (entre 3 y < 10 percentil), eutrófico (entre 10 y < 90 percentil), sobrepeso (entre 90 y 97 percentil) y obeso (> 97 percentil). Se registraron todas las manifestaciones clínicas que conformaron la sospecha diagnóstica. El resultado de los exámenes radiográficos simples de tórax se reportaron como: normales, y con hiperinsuflación o con presencia de atelectasias y/o neumonías, de acuerdo con informe radiográfico. Se registró, además, la mutación identificada y el número de menores de un año fallecidos en el periodo del estudio.
Los resultados se analizaron mediante estadística descriptiva en forma de porcentajes. El Comité de Ética del Hospital Pediátrico aprobó la investigación.
RESULTADOS
Se diagnosticaron 44 niños fibroquísticos en el periodo, de los cuales 35 correspondieron a formas con enfermedad pulmonar típica (tabla). En 16 el diagnóstico se realizó antes del año de edad, para 36,4 %. Aunque se diagnostican más casos en el periodo 1993-2003, en el periodo siguiente 2004-2013 hay proporcionalmente un diagnóstico un poco mas temprano, en el primer año de la vida (8/24, 33,3 % y 8/20-40 % respectivamente).
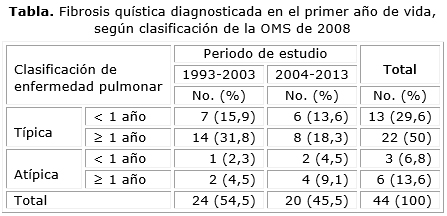
Los niños diagnosticados en la etapa de lactante tuvieron una edad media al diagnóstico de 4,9 meses, y el 68,7 % de ellos se diagnosticaron antes de los 6 meses de edad, con predominio de los varones (Fig. 1).
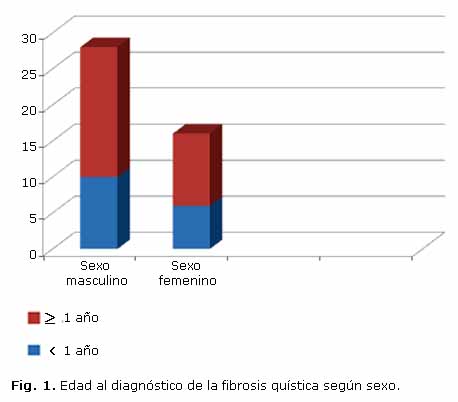
La desnutrición proteico energética estuvo presente en más de la mitad de los menores de un año (Fig. 2), aunque solo 2 tuvieron formas graves asociadas a edema nutricional, ambos en el último periodo de estudio. Las manifestaciones respiratorias, como la polipnea, el tiraje y el tórax hiperinsuflado (Figs. 3 y 4) predominaron, mientras que la distensión abdominal y la esteatorrea fueron las más características dentro de la afectación digestiva. Solo 12 de los 16 lactantes (75 %) presentaban insuficiencia pancreática, todos ellos con formas típicas. No se reportaron enfermos con historia de íleo meconial.
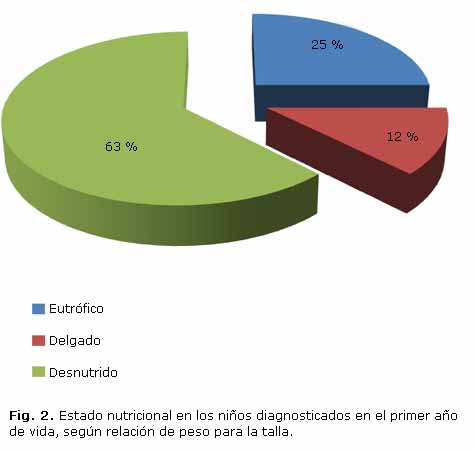
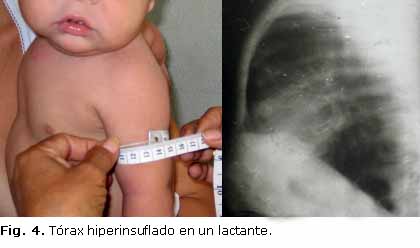
Las radiografías de tórax mostraron hiperinsuflación en todos los casos, intermitente o persistente. Los 13 menores de un año con formas típicas presentaron, al menos, un episodio de atelectasia y/o neumonía.
Se aisló la Pseudomona aeruginosa en 12 niños (75 %). Staphilococcus aureus fue el otro germen aislado en 2 casos; en uno de ellos concomitó con Pseudomona aeruginosa. En 3 pacientes no se aislaron gérmenes.
En los pacientes con estudio genético predominó la mutación delta F 508 (F508del) (31,3 %).
Fallecieron 3 pacientes durante el primer año (18,8 %), por complicaciones infecciosas asociadas a desnutrición severa, todos en el periodo 1993-2003.
DISCUSIÓN
Aunque la FQ es una enfermedad multisistémica, la letalidad depende del compromiso respiratorio, de ahí que es fundamental que el clínico sospeche lo más precozmente posible su diagnóstico, y lo incluya en el diagnóstico diferencial de cualquier cuadro respiratorio recurrente o persistente en los niños pequeños.4
En Argentina, en 2009, D'Alessandro5 comparó 21 niños con diagnóstico por pesquisa con igual número diagnosticados por sospecha clínica, con diferencia significativa en la edad al diagnóstico: 0,16 ± 0,13/1,03 ± 1,23 años respectivamente. También fue mejor y significativo el estado nutricional de acuerdo con el índice de masa corporal (IMC): 0,19 ± 1,04/0,17 ± 0,09.
En niños que presentan íleo meconial al nacimiento (manifestación digestiva determinada genéticamente9 e infrecuente en Cuba),6 o tenían familiares con FQ (antecedente también poco frecuente, dado el carácter autosómico recesivo de esta afección),2,6 es lógico sospechar FQ cuando aparecen manifestaciones respiratorias recurrentes, tanto de sibilancia, tos quintosa o neumonía. No sucede así en el resto de los enfermos, que son la mayoría, por lo que es muy difícil que se realicen diagnósticos antes de los 6 meses de edad.4,10
En este estudio, sin embargo, hay un grupo importante de niños diagnosticados en el primer año de la vida, y la mayoría de ellos antes de los 6 meses, con tendencia al incremento en los últimos 10 años, lo cual puede estar en relación con el trabajo sistemático y la capacitación a los miembros de estos grupos multidisciplinarios.
Estos resultados son algo inferiores a los reportados por Razón y otros6 en Cuba, en 2009, en el estudio de los 234 niños fibroquísticos registrados en el país: 2001-2008, con 49,5 % de diagnóstico antes del año de edad, aunque en el 13,4 % el diagnóstico se realizó a partir de los 16 años. En Santiago de Cuba, en el oriente del país, Guzmán y otros,11 en 2008, reportan que el 41,2 % de sus diagnósticos se hacen antes de los 5 años de edad.
La tomografía computarizada de alta resolución (TACAR) ha permitido demostrar alteraciones pulmonares a edad tan temprana como 3 meses (dilataciones bronquiales y atrapamiento de aire).12-14 Estas alteraciones estructurales, que ocurren en los primeros meses, constituyen un fenómeno de remodelación que conduce a la disminución de la función pulmonar.3
Estudios de lavado broncoalveolar también ponen de manifiesto importante actividad de elastasa libre de los polimorfonucleares neutrófilos, con niveles altos de interleucina 8 (IL-8), como expresión del fenómeno inflamatorio persistente detectado desde el nacimiento.1,15 No es de extrañar, por tanto, que las manifestaciones respiratorias aparezcan muy temprano con episodios de sibilancia persistente, bronquitis y tos crónica, así como neumonía, entre otras.13,16
Es llamativo que 5 lactantes presentaron pérdida salina importante con deshidratación, que no fueron reconocidas de inicio como pérdidas por la piel en el curso de la FQ. Las pérdidas salinas profusas, hasta de 80-100 mg de Na son frecuentes en climas cálidos,9 como el de Cuba. La hipoelectrolitemia puede ser crónica, y se caracteriza por anorexia, irritabilidad, vómitos y fallo en el crecimiento, los que pueden atribuirse erróneamente a fenómenos infecciosos.2,9
La insuficiencia pancreática es característica de enfermos con mutaciones clase I a III, y puede afectar hasta el 85-90 % de estos.9,14 La más frecuente de estas mutaciones es la F508del, de la clase II,17 que también es la más frecuente en Cuba (37 %),2,6 y en nuestra muestra representó el 31,3 %. Para la afectación respiratoria existe pobre correlación del genotipo con el fenotipo, debido a genes modificadores y otros factores.2,8,17
La insuficiencia pancreática aparece cuando se ha perdido el 95 % de la función exocrina pancreática, lo que resulta en malabsorción de proteínas, grasas, y en menor medida, de carbohidratos.9
Algunos síntomas, como el prolapso rectal, descritos hasta en el 20 % de los niños entre 6 meses y 3 años, y las formas nutricionales graves con edema, hepatomegalia y lesiones en piel, que aparecen habitualmente en los primeros 6 meses, no se describen en los diagnosticados mediante pesquisa.9,18 En la muestra estudiada los 2 niños con la forma clínica nutricional severa evolucionaron satisfactoriamente, aunque la mortalidad es elevada en estos casos. La buena nutrición mejora, a su vez, el pronóstico de la enfermedad.4
Las formas leves, no típicas, generalmente se diagnostican más tarde.7
La colonización por bacterias patógenas, fundamentalmente Pseudomonas y S. aureus, puede ocurrir tempranamente en estos casos, hasta en el 15-30 % de los lactantes.19 Se plantea que la colonización es más frecuente en niños que presentaron íleo meconial, cuando se utiliza aerosolterapia, en pacientes homocigotos a la mutación delta F 508, en hospitalizaciones frecuentes, en hijos de madres con pobre conocimiento de la enfermedad, y si tienen hermanos mayores fibroquísticos, entre otros factores.4,8,19 Aunque predominan en la muestra estudiada los varones, al igual que en otros reportes,2,6,11,13 sin embargo, se considera el sexo femenino como el de más riesgo de colonización.19 La colonización con Pseudomonas es un factor de mal pronóstico,4 y se considera un marcador de la enfermedad.19
Después de la aparición inicial de esta bacteria de forma intermitente en las muestras obtenidas de las vías aéreas, si no se trata agresivamente, ocurre un crecimiento excesivo de la población bacteriana, y se produce un cambio al fenotipo mucoide; se forma un biofilm que perpetúa la infección. Este fenómeno es responsable del rápido deterioro clínico, la disminución de la función pulmonar y la muerte.4,7,16,19-21 En las exacerbaciones respiratorias que sufren estos pacientes juegan un rol importante las Pseudomonas, seguido por los virus respiratorios y otras bacterias.21
La hiperinsuflación es un hallazgo precoz,12,14 y puede detectarse por radiografía simple, ya que la TACAR, aunque es más sensitiva para detectar progresión de la enfermedad pulmonar,3,14 es más costosa y expone a mayor cantidad de radiaciones.16 Las bronquiectasias detectadas en los primeros meses pueden tener carácter transitorio, aunque la tercera parte de ellas persiste, y en un grupo similar pueden aparecer posteriormente.12 Se plantea que constituyen factores de riesgo para bronquiectasias el íleo meconial, los síntomas respiratorios persistentes, la actividad incrementada de elastasa de los neutrófilos libre en el líquido de lavado broncoalveolar, así como la presencia de atrapamiento de aire en la radiografía simple de tórax.15
En 170 lactantes con una edad media de 3,7 meses, Wainwright13 describe, mediante TACAR, bronquiectasias en el 58 %, y atrapamiento de aire en el 45 %. No son sorprendentes estos resultados, ya que refieren 68 % de niños homocigotos para la mutación F508del, con síntomas de tos y sibilancias muy tempranas e historia de íleo meconial en el 20 %.
La mortalidad se plantea que es mayor en las clases pobres, y cuando el enfermo está expuesto al tabaquismo u otros polutantes. También se incrementa cuando la infección por Pseudomonas se cronifica, si no hay adherencia al tratamiento, y si la ingestión de calorías no cubre los requerimientos incrementados en estos pacientes.7
Para Cuba, en el periodo 2001-2008, Razón6 describe un promedio anual de fallecidos de 3,75, de ellos el 33,3 % ocurre en los menores de un año.
Cuba tiene un trabajo sostenido en el seguimiento multidisciplinario de estos enfermos. El estado cubano garantiza, de forma gratuita, los medicamentos fundamentales para el tratamiento de esta enfermedad, así como una dieta diferenciada que garantice el aporte nutricional necesario. Cada año se realizan Talleres Nacionales para el intercambio entre los grupos de trabajo y actividades de capacitación y entrenamiento en técnicas de fisioterapia respiratoria. Por otro lado, se ha dado solución a un importante grupo de necesidades socioeconómicas que tenían algunos pacientes y que repercutían negativamente en su calidad de vida.6
No hay dudas de que hay que seguir trabajando por elevar al máximo la sospecha clínica en niños con manifestaciones respiratorias persistentes y desnutrición en etapas tempranas de la vida. Es recomendable la introducción de la pesquisa neonatal para lograr un diagnóstico más temprano.
REFERENCIAS BIBLIOGRÁFICAS
1. Sly P, Brennan S, Gangell C. Lung disease at diagnosis in infants with Cystic fibrosis detected by newborn screening. J Respir Crit Care Med. 2009;80:146-52.
2. Rojo MJ. Fibrosis quística o mucoviscidosis. En: De la Torre E, Pelayo E, editores. Pediatría.Tomo III. . La Habana: Editorial Ciencias Médicas; 2007. p. 1012-54.
3. Regamey N, Jeffery PK, Alton EWFW, Bush A, Davies JC. Airway remodelling its relationship to inflammation in Cystic fibrosis. Thorax. 2011;66:624-9.
4. Fielbaum O. Avances en Fibrosis quística. Rev Med Clin Condes. 2011;22(2)150-9.
5. D'Alessandro V, Rentería F, Fernández A, Martínez MI, Segal E. Comparación del estado clínico-funcional en niños con fibrosis quística detectados por pesquisa neonatal o por síntomas clínicos. Arch Argent Pediatr. Oct 2009;107(5):430-5.
6. Razón R, Rodríguez F, Rojo MJ, González JA, Abreu G, Pérez T, et al. La fibrosis quística en Cuba. Rev Cubana Pediatr. 2009;81(Supl):85-92.
7. Wolfenden LL, Schechter MS. Genetic and non-genetic determinants of outcomes in Cystic fibrosis. Paediatr Respir Rev. 2009;10:32-6.
8. Slieker MG, van der Berg JMW, Kouwenberg J, Teding van Berkhout F, Heijerman HGM, van der Ent CK. Long-term effects of birth order and age at diagnosis in cystic fibrosis: a sibling cohort study. Pediatr Pulmonol. 2010;45:601-7.
9. Robinson P. Digestive Manifestations in Cystic Fibrosis. In: Taussig LM, Landau LI, editors. Pediatric Respiratory Medicine. 2nd ed. Philadelphia: Mosby Elsevier; 2008. p. 889.
10. Dodge JA, Lewis PA, Stanton M, Wilsher J. Cystic fibrosis mortality and survival in the UK: 1947-2003. Eur Respir J. 2007;29:522-6.
11. Guzmán K, del Campo E, Nápoles N, Toledano G, Coello D. Principales características clínico epidemiológicas de pacientes con fibrosis quística en la provincia de Santiago de Cuba. Medisan. 2011;15(2):152.
12. Mott LS, Park J, Murray CP, Gangell CL, de Klerk NH, Robinson PJ, et al. Progression of early structural lung disease in young children with cystic fibrosis assesedusing CT. Thorax [serie en Internet]. 2011 [citado 13 de abril de 2012]. Disponible en: http://thorax.bmj.com/content/early/2011/12/26/thoraxjnl-2011-200912.full.html
13. Wainwright CE, Vidmar S, Armstrong DS, Byrnes CA, Carlin JB, Cheney J, et al. Effect of bronchioalveolar lavage directed therapy on Pseudomonas aeruginosa infection and structural lung injury in children with Cystic fibrosis: a randomized trial. JAMA. 2011;306(2):163-71.
14. Egan M. Fibrosis Quística. En: Kliegman RM, Stanton BF, editores. Nelson. Tratado de Pediatría. 19th ed. Barcelona: Saunders Elsevier; 2013. p. 1540-56.
15. Sly PD, Gangell CL, Chen L, Ware RS, Ranganathan S, Mott LS, et al. Risk factors for bronchiectasis in children with Cystic fibrosis. NEJM. 2013;368(21):1963-70.
16. Tiddens HAWM, Rosenfeld M. Respiratory manifestations in Cystic fibrosis. En: Taussig LM, Landau LI, editores. Pediatric Respiratory Medicine. 2nd ed. Philadelphia: Mosby Elsevier; 2008. p. 871-87.
17. Simmonds NJ, D'Souza L, Roughton M, Alton EWFW, Davies JC, Hodson ME. Cystic fibrosis and survival to 40 years: a study of Cystic fibrosis transmembrane conductance regulator function. Eur Respir J. 2011;37:1076-82.
18. Muniz AE, Bartle S, Foster R. Edema, anemia and acrodermatitis enteropathica: an uncommon initial presentation of Cystic fibrosis. Pediatr Emerg Care. 2004;20(2):112-4.
19. Levy H, Kalish LA, Cannon CL, Garcia KC, Gerard C, Goldman D, et al. Predictors of mucoid pseudomonas colonization in Cystic Fibrosis patients. Pediatr Pulmonol. 2008;43:463-71.
20. Callagham M, Mc Clean S. Bacterial host interactions in Cystic Fibrosis. Curr Opinion Microbiol. 2012;15:71-7.
21. Stenbit AE, Flume PA. Pulmonary exacerbations in Cystic fibrosis. Curr Opinion Microbiol. 2011;17:442-7.
Recibido: 8 de abril de 2014.
Aprobado: 21 de abril de 2014.
Gladys Fuentes Fernández. Hospital Pediátrico Centro Habana. Calle Benjumeda y Morales, municipio Cerro. La Habana, Cuba. Correo electrónico: gff@infomed.sld.cu
