










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Pediatría
versión impresa ISSN 0034-7531
Rev Cubana Pediatr vol.86 no.4 Ciudad de la Habana oct.-dic. 2014
ARTÍCULO DE REVISIÓN
Enfermedades pulmonares intersticiales en el niño
Interstitial pulmonary diseases seen in the child
Dr.C. Roberto Razón Behar, MSc. Daisy Hevia Bernal
Hospital Pediátrico Universitario "William Soler". La Habana, Cuba.
RESUMEN
Las enfermedades pulmonares intersticiales en el niño, comprenden un gran y heterogéneo grupo de raras enfermedades difusas pulmonares de morbilidad variada. Se caracterizan por cambios inflamatorios y fibróticos que causan remodelación de las paredes alveolares y de las vías respiratorias distales, y ocasionan un defecto restrictivo funcional y una alteración en el intercambio gaseoso, con hipoxemia progresiva. Son muchas las enfermedades que pueden afectar al intersticio pulmonar y múltiples las formas etiológicas causadas por una variedad de procesos patológicos, que incluyen, factores genéticos, asociación a enfermedades sistémicas, así como a respuestas inflamatorias o fibróticas a diferentes estímulos. Sin embargo, algunas veces su origen es desconocido, y se catalogan entonces como idiopáticas. Los neumólogos pediátricos han tratado de clasificar los casos de las enfermedades pulmonares intersticiales en las diferentes categorías descritas originalmente en adultos, pero, en realidad, existen formas del adulto que no se observan en la infancia, y formas únicas de presentación pediátrica. Se señala la importancia del conocimiento de estas entidades, particularmente las de origen desconocido o idiopáticas.
Palabras clave: enfermedades pulmonares intersticiales, neumonías intersticiales idiopáticas, fibrosis pulmonar.
ABSTRACT
The interstitial pulmonary diseases seen in the child comprise a large heterogeneous group of rare diffuse pulmonary diseases of varied morbidity. They are characterized by inflammatory and fibrotic changes causing remodeling of alveolar walls and of the distal respiratory pathways, leading to restrictive functional defect and altered gas exchange with progressive hypoxemia. Many diseases can affect the pulmonary insterstice and the etiological forms caused by a variety of pathological processes are multiple including genetic factors, association to systemic diseases and inflammatory or fibrotic responses to different stimuli. However, the origin is sometimes unknown, so they are classified as idiopathic diseases. The pediatric pneumologists have tried to classify the interstitial pulmonary disease cases into the originally described categories for adults; however, there are adult forms that do not occur in childhood and unique pediatric presentations that are not seen in adulthood. The importance of knowledge about these diseases, particularly those of unknown or idiopathic origin was stressed in this article.
Keywords: interstitial pulmonary diseases, idiopathic interstitial pneumonias, pulmonary fibrosis.
INTRODUCCIÓN
Las enfermedades pulmonares intersticialesen el niño (EPI, en idioma inglés interstitial lung disease [ILD] in children [chILD]), comprenden un gran y heterogéneo grupo de raras enfermedades difusas pulmonares de morbilidad variada. Se caracterizan por cambios inflamatorios y fibróticos que causan remodelación de las paredes alveolares y de las vías respiratorias distales, y ocasionan un defecto restrictivo funcional y una alteración en el intercambio gaseoso con hipoxemia progresiva. Los niños afectados presentan síntomas y signos de dificultad respiratoria, taquipnea, estertores y alteraciones, radiográficas torácicas, caracterizadas por la presencia de infiltrados difusos.1,2
Son muchas las enfermedades que pueden afectar al intersticio pulmonar y múltiples las formas etiológicas causadas por una variedad de procesos, que incluyen, factores genéticos, asociación a enfermedades sistémicas y a respuestas inflamatorias o fibróticas a diferentes estímulos.2 Sin embargo, algunas veces su origen es desconocido, y se catalogan entonces como idiopáticas.1
Las EPI en niños son muy diferentes en muchos aspectos a las del adulto. En primer lugar, son raras (estimadas en 0,36-1,32 por cada 100 000 habitantes en niños, comparado con 60-80 por 100 000 en adultos).3 En segundo lugar, el espectro de condiciones, particularmente en los menores de 2 años, es mucho más amplio que en los adultos, lo cual está relacionado con el hecho de que estas enfermedades ocurren en el contexto del crecimiento pulmonar y de los diferentes estadios de la maduración y desarrollo alveolar, cada uno de los cuales son regulados por eventos específicos en cascada.4
En los niños las EPI son más frecuentemente diagnosticadas en el primer año de la vida, particularmente ciertas entidades como la glucogenosis pulmonar intersticial, la hiperplasia de células neuroendocrinas y las alteraciones genéticas de metabolismo del surfactante. En niños mayores la patogénesis de las EPI comparten similitudes con los procesos que se observan en adultos, y los cambios patológicos se caracterizan por alteraciones de los septum alveolares con la presencia de zonas focales de proliferación fibroblástica. Estas lesiones de parches fibróticos aparecen en sitios de daños alveolares recientes, y su número se relaciona con el empeoramiento de la función pulmonar y con un peor pronóstico.4
Nuestros objetivos principales son revisar las clasificaciones y la fisiopatología de estas entidades, y profundizar en el conocimiento de las EPI idiopáticas.
DESARROLLO
Clasificación
La neumonitis intersticial aguda fue descrita por primera vez en 1935 por Louis Hammany Arnold Rich.5 Estos autores, en 1944,describieron 4 pacientes que presentaban procesos respiratorios crónicos con evolución desfavorable, en los que se observaban lesiones intersticiales pulmonares que producían cicatrices extensas. Ellos no señalaron ningún agente etiológico. Esta entidad recibió el nombre de síndrome de Hamman-Rich.6
Scadding, en 1967, señaló el término de alveolitis fibrosante mural, cuando el proceso se caracterizaba por el engrosamiento de los tabiques alveolares con células mononucleares en el intersticio, y el término de alveolitis fibrosante descamativa, si había células descamativas en el interior de los alvéolos.7
Como resultado de la rareza de las EPI en los niños y las diferencias importantes entre las que afectan la niñez y las que afectan a los adultos, existe una gran confusión en su nomenclatura, clasificación y tratamiento.8 Los neumólogos pediátricos han tratado de clasificar los casos de EPI en las diferentes categorías descritas originalmente en adultos, pero, en realidad, existen formas del adulto que no se observan en la infancia, y formas únicas de presentación pediátrica.9
Liebow y Carrington, en 1969, clasificaron este grupo de entidades en 5 variedades de acuerdo con los componentes alveolares predominantes y las estructuras afectadas: la neumonía intersticial clásica o usual (UIP), la bronquiolitis obliterante (BO), la neumonía intersticial descamativa (DIP), la neumonía intersticial linfoidea y la neumonía intersticial a células gigantes.10
En años posteriores han surgido numerosas propuestas y revisiones de las clasificaciones. En 2002, la Sociedad Torácica Americana y la Sociedad Respiratoria Europea revisaron la clasificación de las neumonías intersticiales idiopáticas.11
Deutsch y otros, en 2007, publican la experiencia de un grupo de hospitales pediátricos norteamericanos en niños menores de 2 años con enfermedades difusas pulmonares, que incluían biopsias, formas de presentación clínica, el consenso en la revisión de los hallazgos patológicos y los datos de la evolución.12 Durante este mismo periodo hubo numerosos avances en el conocimiento de la etiología genética de varias enfermedades pulmonares pediátricas. Un estudio posterior (2008), caracterizó las neumonías intersticiales no específicas (NSIP). Este sistema de clasificación enfatiza en el reconocimiento de los patrones patológicos y su correlación con los hallazgos clínicos y radiológicos, para establecer un diagnóstico final clínico-radiológico-patológico.13
En 2013, la Sociedad Torácica Americana publicó una guía práctica para la clasificación, evaluación y tratamiento de las EPI del niño menor de 2 años,14 basada en la clasificación descrita por Deutsch y otros12 (anexo).
En la clasificación en menores de 2 años, estas entidades se han categorizado basados en los hallazgos histopatológicos en: desórdenes difusos del desarrollo, anomalías en el crecimiento -lo que refleja una alveolarización deficiente-, desórdenes específicos de etiología no definida como la hiperplasia de células neuroendocrinas de la infancia y la glucogenosis intersticial pulmonar, que son 2 entidades distintas que se presentan tempranamente en la vida y que tienen características particulares. Ambas tienen un pronóstico favorable.2,15
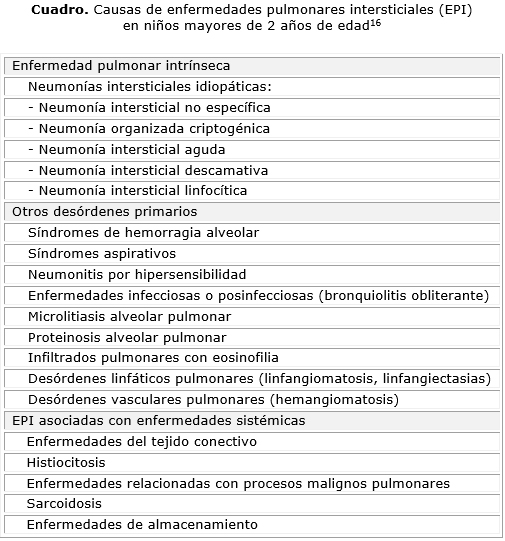
La clasificación de la EPI en niños de 2 a 16 años, está basada en los esquemas de adultos y está menos definida. Las enfermedades mediadas por la inmunidad, las enfermedades sistémicas y las enfermedades infecciosas y posinfecciosas son las más frecuentes. Existe un gran número de condiciones genéticas raras, que pueden también presentarse en los adultos; la misma enfermedad genética puede causar diferentes aspectos histológicos, que pueden estar relacionados con la edad.3 Vece y Fan,16 en 2010, publicaron una clasificación de las EPI en niños mayores de 2 años (cuadro).
Fisiopatología
En las neumopatías intersticiales crónicas, por causas desconocidas, o por la presencia de un agente infeccioso, un tóxico, una acumulación del surfactante C anormal, o un antígeno, pueden provocar una lesión de los neumocitos tipo II. Estas células no cumplen adecuadamente su función de mantener la homeostasis del surfactante pulmonar. Esta afectación inicial, es la fase de alveolitis. La presencia de una alveolitis persistente, daña el pulmón y modula la fibrogénesis. Después de un efecto dañino inicial, comienza la respuesta inflamatoria, que estimula la proliferación de células epiteliales y de células mesenquimatosas activadas y la producción de la matriz extracelular.4 Esta respuesta usualmente es seguida por la apoptosis de las células de reparación y la producción de la matriz con fibrolisis para asegurar la curación. En el caso de las EPI, el proceso fibrótico patológico no parece vinculado a una respuesta inflamatoria consecutiva y crónica, pero sí a la continuación de la producción de la matriz con una reducción de su movilización y una apoptosis alterada de las células alveolares.17
La respuesta inflamatoria crónica perpetúa el reclutamiento de las células inflamatorias e inmunorreguladoras en el intersticio, paredes alveolares y tejidos perialveolares, y conduce al engrosamiento de las paredes alveolares con fibrosis extensa y pérdida de la función del intercambio gaseoso alveolar.4
El patrón patológico puede ser el resultado de la reparación anormal del pulmón; el interés, por lo tanto, está enfocado en el daño de la superficie alveolar con disrupción marcada de la integridad del epitelio durante el curso de la enfermedad y la acumulación y activación de células inmunoinflamatorias, con la subsecuente migración y proliferación de los miofibroblastos y el depósito de la matriz extracelular.18
Un paso importante en este proceso es la capacidad de los neumocitos alveolares tipo II de iniciar rápidamente la reepitelización. Estas células representan a la célula madre del epitelio alveolar, dada su habilidad para proliferar y continuar a la transición terminal en neumocitos tipo I. En situaciones de daño extenso de la superficie pulmonar hay una demora en la iniciación y progreso del proceso de reepitelización.19 Como consecuencia, la denudación prolongada de la membrana basal puede contribuir a interacciones entre las células alveolares epiteliales y las células mesenquimatosas, y ocasionar profundas modificaciones de las funciones celulares con un desbalance en la producción de mediadores polipéptidos, incluyendo citoquinas, factores de crecimiento, proteasas y oxidantes. La población local de fibroblastos y miofibroblastos, puede aumentar progresivamente por la estimulación y proliferación de factores mitogénicos y reducción de la apoptosis. Ello lleva a una remodelación aberrante y progresiva de los tejidos con depósitos de los componentes de la matriz desorganizada, incluyendo fibras colágenas, fibras elásticas, fibronectina y proteoglicanos.20
Una cascada de mediadores que se producen localmente por las células inflamatorias mensenquimatosas y epiteliales, juegan un papel crítico en la progresión de los cambios fibróticos.21
Recientes hallazgos genéticos han identificado mutaciones causales en los genes de la proteína del surfactante (PS), fundamentalmente los genes de la PS-B y PS-C, y de otros genes incluidos en el metabolismo del surfactante, como el ABCA3.22
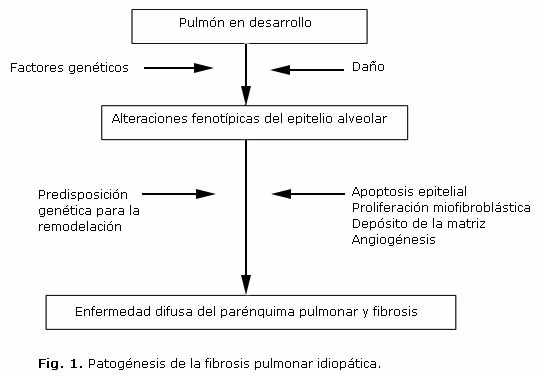
Adicionalmente, la arquitectura anormal pulmonar que se observa en la fibrosis pulmonar está asociada a la formación de nuevos vasos sanguíneos. Este proceso requiere la secreción de moléculas angiogénicas que promuevan la migración celular endotelial y la neovascularización4 (Fig. 1).
Neumonías intersticiales idiopáticas
La clasificación actual de las neumonías intersticiales idiopáticas en adultos se basa en una declaración de consenso de un grupo multidisciplinario de neumólogos, radiólogos y patólogos encargados por las Sociedades Torácica Americana y la Respiratoria Europea.11 El panel reconoció 7 entidades clínico-radiológicas-patológicas: la fibrosis pulmonar idiopática (IPF), la NSIP, la neumonía organizada criptogénica (COP), la enfermedad pulmonar intersticial asociada con bronquiolitis respiratoria (RB-ILD), la neumonía intersticial aguda (AIP), la DIP y la neumonía intersticial linfoide (LIP). Con la excepción de la IPF y la RB-ILD, las neumonías intersticiales idiopáticas restantes se pueden observar en niños, aunque son muy raras.16
La fibrosis pulmonar idiopática, la forma más común de la neumonía intersticial idiopática en adultos, es un trastorno progresivo y fatal caracterizado patológicamente por neumonía intersticial clásica o usual (UIP), una lesión heterogénea de fibrosis desigual y en panal de abeja, alternando zonas de leve a moderada inflamación con pulmón normal. La identificación de focos fibroblásticos característicos, es necesaria para el diagnóstico. Aunque muchos niños se han reportado erróneamente como IPF, este patrón solo se ha descrito en un adolescente con mutaciones ABCA3.23
Como en adultos, la neumonía intersticial inespecífica en niños, se considera una forma distintiva de las EPI, que tienen un mejor pronóstico que la IPF. Histológicamente está caracterizada por una mezcla de inflamación y fibrosis, y subdividido en un patrón celular y un patrón fibrosante, basado en la importancia relativa de estos componentes en particular. Aunque la mayoría de los casos es de etiología desconocida, se han descrito en errores innatos del metabolismo del surfactante, trastornos autoinmunitarios y neumonitis por hipersensibilidad. El patrón celular tiene una mejor respuesta a la terapia esteroide y se puede resolver completamente, mientras que el patrón fibrótico es más probable que progrese a una enfermedad pulmonar terminal.16
La COP, anteriormente llamada bronquiolitis obliterante, se ha descrito en niños como un fenómeno aislado o con asma, reacciones a medicamentos, infecciones, procesos malignos sometidos a quimioterapia, trasplantes de médula ósea y procesos autoinmunes. El pronóstico es generalmente bueno, con una respuesta favorable al tratamiento con corticosteroides.16
La neumonía intersticial aguda ocurre raramente en niños. El patrón histológico de esta entidad clínica es el daño alveolar difuso. Tiene una evolución rápidamente progresiva, y a menudo, fatal. Los corticosteroides y otros tratamientos generalmente son ineficaces.16
La DIP, también se ha presentado en niños, pero en contraste con la de los adultos, no está relacionada con el hábito de fumar. Actualmente se reconoce que la DIP en niños, casi siempre es causada por un error innato del metabolismo del surfactante. Histológicamente no presenta necrosis alveolar, exudado, ni membrana hialina; la característica más notable es la proliferación y descamación de neumocitos II dentro de los alvéolos, y engrosamiento de las paredes alveolares, con menor cantidad de infiltrado intersticial de linfocitos y células plasmáticas. La DIP en los niños, particularmente en los menores de 2 años, tiene un mal pronóstico.16
Aunque está incluida como una de las neumonías intersticiales idiopáticas, la neumonía intersticial linfocítica es una forma de un trastorno linfo-proliferativo, que se observa más a menudo en pacientes con enfermedades del tejido conectivo o en inmunodeficiencias. Anteriormente, la manifestación pulmonar más común del niño con Sida, era la LIP. Hoy en día se observa con mucho menos frecuencia debido al desarrollo del tratamiento de las infecciones por el VIH. Se caracteriza por la afectación casi exclusiva del espacio intersticial con infiltración linfocitaria y de células plasmáticas.16
Diagnóstico
El cuadro clínico se caracteriza por la presencia de insuficiencia respiratoria progresiva, debido a la fibrosis intersticial. El comienzo es insidioso, con tos seca e irritativa, y la presencia de disnea, inicialmente durante el esfuerzo, pero, posteriormente en reposo. Puede evolucionar en meses o años después hacia la insuficiencia respiratoria crónica. En fases más avanzadas presentan, además, cianosis, anorexia, fatigas, pérdida de peso y dolor torácico, a veces de origen pleural. El neumotórax puede ocurrir en el 30 % de los casos.24,25
En el examen físico puede encontrarse taquicardia, taquipnea, cianosis, estertores crepitantes y subcrepitantes finos, más localizados hacia las bases pulmonares, uñas "en vidrio de reloj" y dedos hipocráticos. La fiebre es inusual, pero cuando se presenta puede haber una infección neumónica sobreañadida, la cual puede ser la forma de presentación de la enfermedad, y también representar un serio problema para la vida del paciente.24
En el periodo terminal se pueden observar signos hipertensión pulmonar y falla cardíaca ventricular derecha. La presencia de hipertensión pulmonar es considerada como riesgo importante de mortalidad en más de 30 %.24
Las radiografías torácicas muestran en fases tempranas de la enfermedad, imágenes borrosas hacia las bases con aspecto de vidrio deslustrado. Si la lesión avanza, se puede apreciar un patrón difuso reticular, micronodular o mixto, que se irradia desde los hilios a lo largo de los bordes del corazón hacia las bases. En los niños las imágenes pueden tener una característica más irregular. En las fases más avanzadas se observa el patrón de "panal de abeja".24,25
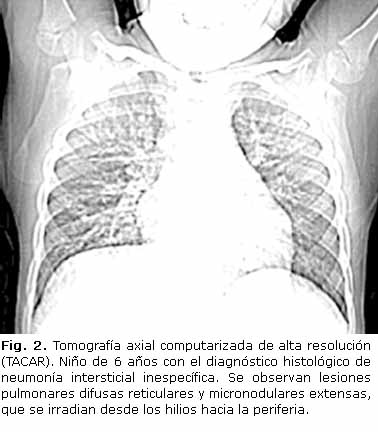
La tomografía axial computarizada de alta resolución (TACAR) tiene gran sensibilidad y precisión para detectar las características y la extensión de las lesiones (Fig. 2 y 3).24,25

Las pruebas de función pulmonar y la oximetría de pulso se pueden utilizar para evaluar la severidad de la enfermedad.16,24
El estudio de la función respiratoria ofrece un patrón clásico restrictivo, con disminución de la capacidad vital funcional, la distensibilidad y la capacidad de difusión.24
En niños que, de acuerdo con su edad puedan cooperar, la prueba del ejercicio o caminar por 6 minutos, puede ser un indicador objetivo de la progresión de la enfermedad.16 Se observa con frecuencia hipoxemia que se incrementa con el ejercicio. La presión parcial de anhídrido carbónico (PCO2) está usualmente baja o normal.8,24
No hay alteraciones de laboratorio específicas para esta enfermedad. Sin embargo -aun en ausencia de signos o síntomas característicos de enfermedades del tejido conectivo- se deben realizar determinaciones serológicas de autoinmunidad en todos los pacientes. Más del 90 % tiene una eritrosedimentación elevada. A veces se observa hipergammaglobulinemia, factor reumatoideo y anticuerpos antinucleares positivos, y la presencia de crioglobulinas séricas e inmunocomplejos circulantes.8,24
La gammagrafía con galio o tecnesio se utiliza para detectar los sitios de inflamación y fibrosis, y valorar la evolución del proceso.8,24
Los estudios citológicos del líquido de los lavados broncoalveolares, inicialmente fueron muy prometedores para el diagnóstico de las EPI, pero ahora se consideran como un método más en la investigación, aunque su papel clínico no está claramente establecido. En casos determinados, puede ser de gran ayuda en el diagnóstico diferencial con otras entidades.8,25
En las EPI, el lavado se puede también utilizar para determinar la actividad inflamatoria (porcentaje de linfocitos, neutrófilos, eosinófilos y macrófagos), y la respuesta al tratamiento y su pronóstico. Los estudios seriados por este método tienen cierta utilidad práctica.24
La biopsia pulmonar "a cielo abierto" o por toracoscopia asistida por video, sigue siendo el estándar de oro para el diagnóstico del niño. Se debe interpretar en el contexto de los resultados clínicos y radiológicos.8,16,25
Tratamiento
Es necesario que el tratamiento tenga un enfoque multidisciplinario que incluya al médico de atención primaria, el neumólogo, el nutricionista, el trabajador social, la enfermera y el fisioterapeuta. El tratamiento es sintomático y de soporte, con hincapié en la prevención de las infecciones por influenza y otras prevenibles con vacunas, particularmente, contra el neumococo, y en el tratamiento de las infecciones respiratorias. Es importante también el control ambiental, evitar el humo del tabaco y otros irritantes, participar en programas de rehabilitación pulmonar y realizar regularmente ejercicios físicos.16,24-26
El reflujo gastroesofágico es un factor de riesgo para la aspiración, causa reconocida de neumonitis, por lo que podría contribuir a la inflamación crónica de las vías aéreas y también a la fibrosis, por lo que es importante su diagnóstico y tratamiento.27
Muchos niños en fase avanzada de la enfermedad, requieren oxígeno suplementario durante la noche o continuamente. El desarrollo de la hipertensión pulmonar se asocia con una menor probabilidad de supervivencia, y deben recibir oxígeno y terapia con un vasodilatador pulmonar, como el sildenafil.8,15,16,25,26
Como la inflamación juega un papel importante en muchas formas de EPI en el niño, los corticosteroides -con sus propiedades multifacéticas antiinflamatorias- se consideran la terapia de primera línea. Teniendo en cuenta la mejoría subjetiva en un grupo de pacientes con EPI, y, dado que estas drogas tienen cierta efectividad, se recomienda el tratamiento con corticosteroides.8,15,16,24-27 Se señalan mejores resultados en las variedades UIP y DIP.8
La dosis debe ser alta y mantenida por períodos largos, y disminuirlas lentamente de acuerdo con la respuesta clínica. Dependiendo de la severidad se puede comenzar por vía oral o en pulsos endovenosos. Las dosis diarias orales de prednisona o prednisolona deben ser de 1-2 mg/kg/día, o dosis de pulsos endovenosos de metilprednisolona (10-30 mg/kg), con un máximo de 1 g por 3 días consecutivos cada mes, durante 6 meses.16,24
En pacientes en los que no hubo respuesta anterior a los glucorticoides, se reporta mejoría con la administración del interferón gamma-1b asociado a la prednisolona en dosis bajas. Se supone que el interferón gamma-1b puede influir en el curso de la fibrosis pulmonar idiopática, debido a sus efectos antifibróticos, antiinflamatorios o antiinfecciosos.24 Otros agentes, tales como, la hidroxicloroquina, la azatioprina, el metotrexate, la ciclofosfamida, el meclofenamato y dosis de inmunoglobulina IV, se han utilizado en niños con éxito variable, pero los resultados no son definitivos.8,15,16,24,26,27
La N-acetilcisteína (NAC) aumenta la síntesis de glutatión, y es un potente mediador antioxidante, que disminuye la respuesta fibrótica en modelos animales con fibrosis pulmonar. Diferentes ensayos se han llevado acabo con esta droga sola, o unida a glucocorticoides y azatioprina. Los estudios han demostrado mayor mortalidad e ingresos hospitalarios en los pacientes que recibían la triple terapia, en comparación con el placebo o con el tratamiento con NAC. Por tanto, no se aconseja utilizar esta triple terapia. En la actualidad el ensayo continúa con solo 2 ramas: NAC y placebo. Hasta que se conozcan los resultados, no se podrá dilucidar la verdadera eficacia de la NAC como monoterapia en el tratamiento de la EPI.26,27
Otros fármacos con propiedades antiinflamatorias y antifibróticas, como la pirfenidona, están en diferentes etapas de investigación y aplicación.26,27
Shulgina y otros publicaron un ensayo clínico en el que en un grupo de 181 pacientes con EPI fueron tratados con cotrimoxazol o placebo, y observaron que la adición de cotrimoxazol al tratamiento estándar no tuvo efecto sobre la función pulmonar, pero sí una mejoría en la calidad de vida, y una reducción en la mortalidad por todas las causas en aquellos que tomaron el medicamento.28
El trasplante pulmonar es actualmente el único tratamiento que ha demostrado prolongar la supervivencia y mejorar la calidad de vida de los pacientes con EPI.26,27 Es una opción para el niño que progresa a insuficiencia pulmonar. Los resultados son comparables con los trasplantes para la fibrosis quística o hipertensión pulmonar.16,24
Actualmente, tanto la terapia celular como la terapia génica en la EPI, están en fase de estudio experimental, por lo que todavía se deberá esperar algún tiempo para resolver algunas cuestiones que incluyen aspectos éticos, la seguridad del trasplante de células, las vías ideales para su administración, las indicaciones, su efectividad como tratamiento alternativo o asociado a los tratamientos farmacológicos, y otros aspectos importantes que puedan conducir a conclusiones satisfactorias de la terapia celular.27,29
Evolución y pronóstico
Las EPI, por lo general, tienen una evolución invariablemente progresiva, que está relacionada con la afectación cada vez mayor del tejido pulmonar. La evolución es fatal, y se puede extender desde meses, hasta 1, 2 o más años.8,24
En algunos niños se puede observar una resolución espontánea sin tratamiento, mientras que otros progresan hacia la muerte a pesar de este.24 En la actualidad no existe ningún tratamiento farmacológico que modifique el pronóstico de la enfermedad.26
La hipertensión pulmonar y la falla ventricular derecha son las alteraciones más graves que pueden observarse en la evolución de estas entidades. La mayoría de los niños mueren por fallo respiratorio o cardiorrespiratorio, y el episodio final generalmente está precipitado por infecciones respiratorias intercurrentes.24
Propuesta de clasificación para las enfermedades pulmonares intersticiales en el niño12,14
I. Desórdenes más prevalentes en niños de 0-2 años
A. Desórdenes difusos del desarrollo.
1. Displasia acinar.
2. Displasia alveolar congénita.
3. Displasia alveolo-capilar con alineamiento irregular de las venas pulmonares.
B. Anormalidades del crecimiento.
1. Hipoplasia pulmonar.
2. Enfermedad pulmonar neonatal crónica.
2.1. Enfermedad pulmonar crónica relacionada con la prematuridad (displasia broncopulmonar).
2.2. Enfermedad pulmonar crónica adquirida en lactantes a término.
3. Cambios estructurales pulmonares con anormalidades cromosómicas.
3.1. Trisomía 21.
3.2. Otros.
4. Asociados con enfermedades cardiacas congénitas en niños con cromosomas normales.
C. Condiciones específicas de etiología indefinida.
1. Glucogenosis intersticial pulmonar.
2. Hiperplasia de células neuroendocrinas en la infancia.
D. Desórdenes relacionados con mutaciones genéticas de disfunción del surfactante.
1. Mutaciones (proteína B) SPFTB-proteinosis alveolar pulmonar y variante dominante de patrón histológico.
2. Mutaciones (proteína C) SPFTC-neumonitis crónica de la infancia, patrón dominante histológico; también DIP y NSIP.
3. Mutaciones genéticas ABCA3-proteinosis alveolar pulmonar patrón de variante dominante; también neumonitis crónica de la infancia, DIP y NSIP.
4. Otras con histología consistentes en desórdenes de la disfunción del surfactante sin alteraciones genéticas aún reconocidas.
II. Desórdenes no solo prevalentes en los niños menores de 2 años
A. Desórdenes del huésped normal.
1. Procesos infecciosos y posinfecciosos.
2. Desórdenes relacionados con agentes ambientales: neumonía por hipersensibilidad, inhalación de tóxicos.
3. Síndromes por aspiración.
4. Neumonía eosinofílica.
B. Desórdenes relacionados con enfermedades sistémicas.
1. Desórdenes relacionados con la inmunidad.
2. Enfermedades de almacenamiento.
3. Sarcoidosis.
4. Histiocitosis por células de Langerhans.
5. Infiltrados malignos.
C. Desórdenes del huésped inmunocomprometido.
1. Infecciones oportunistas
2. Desórdenes relacionados con la intervención terapéutica.
3. Desórdenes relacionados a los trasplantes y síndromes de rechazo.
4. Daño difuso alveolar de etiología desconocida.
D. Desórdenes que se asemejan a una enfermedad intersticial.
1. Vasculopatía arterial hipertensiva.
2. Vasculopatía congestiva, incluyendo enfermedad veno-oclusiva.
3. Desórdenes linfáticos.
4. Cambios congestivos relacionados a disfunción cardiaca.
III. No clasificadas (incluyen enfermedades en estado final), biopsias sin un diagnóstico y aquellas con material inadecuado
REFERENCIAS BIBLIOGRÁFICAS
1. Escribano MA, Sirvent JG. Enfermedad pulmonar intersticial en I Curso Nacional de Actualización en Neumología Pediátrica. Madrid: Ergon; 2004. p. 195-213.
2. Das S, Langstonb C, Fan LL. Interstitial lung disease in children. Curr Opin Pediat. 2011;23:325-31.
3. Shailendra Dasa S, Langstonb C, Fan L. Interstitial lung disease in children. Curr Opin Pediat. 2011;23:325-31.
4. Clement A, Eber E. Interstitial lung diseases in infants and children. Eur Respir J. 2008;31:658-66.
5. Hamman L, Rich AR. Fulminating diffuse interstitial fibrosis of the lungs. Transactions of the American Clinical and Climatological Association. European Respiratory Society. 1935;51:154-63.
6. Hamman L, Rich AR. Acute diffuse interstitial fibrosis of the lungs. Bull Johns Hopkins Hosp. 1944;74:177-212.
7. Scadding JG, Hinson KFW. Diffuse fibrosing alveolitis (diffuse interstitial fibrosis of the lungs). Correlation of histology at biopsy with prognosis. Thorax. 1967;22:291-304.
8. Hagood JS, Com G, Vaughan DJ, Young DW, Mroczek-Musulman EC, Young LR, et al. Children's Interstitial Lung Disease (ChILD) Medscape [homepage en Internet] Apr 2; 2012 [citado 17 de febrero de 2014]. Disponible en: http://emedicine.medscape.com/article/1003631-overview#showall
9. Fan LL, Langston C. Pediatric interstitial lung disease. Children are not small adults. Am J Respir Crit Care Med. 2002;165:1466-7.
10. Liebow AA, Carrington CB. The interstitial pneumonias. In: Simon M, Potchen EJ, LeMay M, editors. Frontiers of pulmonary radiology. New York: Grune & Stratton; 1969. p. 102-41.
11. American Thoracic Society/European Respiratory Society. International multidisciplinary consensus classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med. 2002;165:277-304.
12. Deutsch GH, Young LR, Deterding RR, Fan LL, Dell SD, Bean JA, et al. Pathology Cooperative Group; ChILD Research Co-operative. Diffuse lung disease in young children: application of a novel classification scheme. Am J Respir Crit Care Med. 2007;176:1120-8.
13. Travis WD, Hunninghake G, King TE, Lynch DA, Colby TV, Galvin JR, et al. Idiopathic nonspecific interstitial pneumonia: Report of an American Thoracic Society project. Am J Respir Crit Care Med. 2008;177:1338-47.
14. Kurland G, Deterding RR, Hagood JS, Young LR, Brody AS, Castile RG, et al. An Official American Thoracic Society Clinical Practice Guideline: Classification, Evaluation, and Management of Childhood Interstitial Lung Disease in Infancy. Am J Respir Crit Care Med. 2013;188(3):376-94.
15. Bush A, Nicholson AG. Paediatric interstitial lung disease. Eur Respir Mon. 2009;46:319-54.
16. Vece TJ, Fan LL. Interstitial Lung Disease in Children Older Than 2 Years. Pediatr Allergy Immunol Pulmonol. 2010;23(1):33-41.
17. Noble PW, Homer RJ. Back to the future: historical perspective on the pathogenesis of idiopathic pulmonary fibrosis. Am J Respir Cell Mol Biol. 2005;33:113-20.
18. Reynolds HY. Lung inflammation and fibrosis: an alveolar macrophage-centered perspective from the 1970s to 1980s. Am J Respir Crit Care Med. 2005;171:98-102.
19. Barbas-Filho JV, Ferreira MA, Sesso A, Kairalla RA, Carvalho CR, Capelozzi VL. Evidence of type II pneumocyte apoptosis in the pathogenesis of idiopathic pulmonary fibrosis (IFP)/usual interstitial pneumonia (UIP). J Clin Pathol. 2001;54:132-8.
20. Selman M, Pardo A. The epithelial/fibroblastic pathway in the pathogenesis of idiopathic pulmonary fibrosis: tying loose ends. Am J Respir Cell Mol Biol. 2003;29(Suppl. 3):S93-S98.
21. Millar A. Anti-cytokine therapy in fibrosing alveolitis: where are we now? Respir Res. 2000;1(1):3-5.
22. Hartl D, Griese M. Interstitial lung disease in children-genetic background and associated phenotypes. Respir Res. 2005;6:32.
23. Young LR, Nogee LM, Barnett B, Panos RJ, Colby TV, Deutsch GH. Usual interstitial pneumonia in an adolescent with ABCA3 mutations. Chest. 2008;134:192-5.
24. Razón BR. Neumonías intersticiales difusas. En: De la Torre E, González J, Gutiérrez J, Jordán J, Pelayo E. Pediatría. T 3, cap 80. La Habana: Editorial Ciencias Médicas; 2007. p. 1071-4.
25. Deterding RR. Infants and Young Children with Children's Interstitial Lung Disease. Pediatric Allergy, Immunology, and Pulmonology. 2010;23(1):25-31.
26. Marcos PJ, Montero C, Otero GI. Una mirada general a las enfermedades pulmonares intersticiales y una específica a la fibrosis pulmonar idiopática. Galicia Clin. 2011;73(4):13-22.
27. Xaubetab A, Ancocheac J, Bollod E, Fernández-Fabrellase E, Franquetf T, Molina-Molinabg M, et al. Normativa sobre el diagnóstico y tratamiento de la fibrosis pulmonar idiopática. Arch Bronconeumol. 2013;49(8):343-53.
28. Shulgina L, Cahn AP, Chilvers ER, Parfrey H, Clark AB, Wilson ECF, et al. Treating idiopathic pulmonary fibrosis with the addition of co-trimoxazole: a randomised controlledtrial. Thorax. 2013;68:155-62.
29. Moodley Y, Manuelpillai U, Weiss DJ. Cellular therapies for lung disease: a distant horizon. Respirology. 2011;16:223-7.
Recibido: 24 de marzo de 2014.
Aprobado: 14 de abril de 2014.
Roberto Razón Behar. Hospital Pediátrico Universitario "William Soler". San Francisco No. 10112, Reparto Altahabana, municipio Boyeros. La Habana, Cuba. Correo electrónico: roberto.razon@infomed.sld.cu
