










My SciELO
 Custom services
Custom servicesServices on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista Cubana de Pediatría
Print version ISSN 0034-7531On-line version ISSN 1561-3119
Rev Cubana Pediatr vol.87 no.1 Ciudad de la Habana Jan.-Mar. 2015
PRESENTACIÓN DE CASO
Rabdomioma cardíaco
Cardiac rhabdomyoma
MSc. Mariela Céspedes Almira, Dra. Judith Suzarte Portal, MSc. Noel Mansito González
Cardiocentro Pediátrico “William Soler”. La Habana, Cuba.
RESUMEN
El rabdomioma es un tumor benigno cardíaco primario. Se describe un recién nacido de 4 días de edad, producto de parto eutócico a las 40 semanas de edad gestacional y buen peso al nacer, nacido vía transpelviana sin morbilidad perinatal, test de Apgar 9/9, que a las pocas horas de vida se le ausculta soplo cardiaco de intensidad III/VI, sin otros signos clínicos. La evaluación ecocardiográfica reveló masa tumoral pediculada con características obstructivas del tractus de salida del ventrículo izquierdo, que genera gradiente a ese nivel de alrededor de 80 mmHg. Se realizó exéresis del tumor de base ancha, aspecto nacarado y superficie lisa a través de aortotomía oblicua, con circulación extracorpórea. El curso posoperatorio fue sin complicaciones. El paciente evoluciona sin residuos ni secuelas a los 12 meses de seguimiento.
Palabras clave: rabdomioma, cirugía, ecocardiograma.
ABSTRACT
Rhabdomyoma is a primary cardiac benign tumor. This report described a 4 days-old newborn born to eutocic delivery at 40 weeks of gestational age and good birthweight. He was born through transpelvic route with no perinatal morbidity, Apgar test of 9/9, but few hours after birth, he was detected an intensity II/IV cardiac murmur with no other clinical signs. The echocardiographic results showed pedicled tumor mass with obstructive characteristics of the outlet tract of the left ventricle, thus generating around 80 mmHg gradient at that level. This wide-base, pearly and flat surface tumor was excised by means of oblique aortotomy using extracorporeal circulation. The postoperative phase was uncomplicated. The patient recovers with neither residues nor sequelae after 12 months of follow/up.
Keywords: rhabdomyoma, surgery, echocardiogram.
INTRODUCCIÓN
Los tumores cardíacos primarios son poco frecuentes en edades pediátricas. En estudios realizados en niños a través de autopsias se reporta una incidencia de 0,027 a 0,08 %,1,2 mientras que algunos estudios estiman una incidencia de los tumores cardíacos en la vida fetal entre 0,05 a 0,14 %. En la población infantil es más frecuente la presencia de tumores benignos, y es el rabdomioma el tumor detectado en cerca del 90 % de esos casos, al que le siguen los teratomas, fibromas y hemangiomas.3
El primer rabdomioma fue descrito por Von Recklinghausen en 1862. Hasta los años 70 el estudio de elección para su diagnóstico era el angiocardiograma, que se sustituyó posteriormente por el ecocardiograma. En 1982 De Vore hizo el diagnóstico de tumor in útero.4,5
En el estudio de la evolución natural de este tipo de tumor se observa un gran porcentaje de casos con regresión de este desde el final del embarazo y en los primeros años de vida.1 Por lo infrecuente de la entidad se consideró válido reportar este paciente.
PRESENTACIÓN DEL CASO
Se presenta un recién nacido de 4 días de edad, con historia de haber nacido producto de parto eutócico a las 40 semanas de edad gestacional, y peso al nacer de 4 000 g, Apgar 9/9. A las pocas horas de vida se le ausculta soplo cardiaco de intensidad III/VI, sin otros signos clínicos, y se alimentaba con lactancia materna exclusiva.
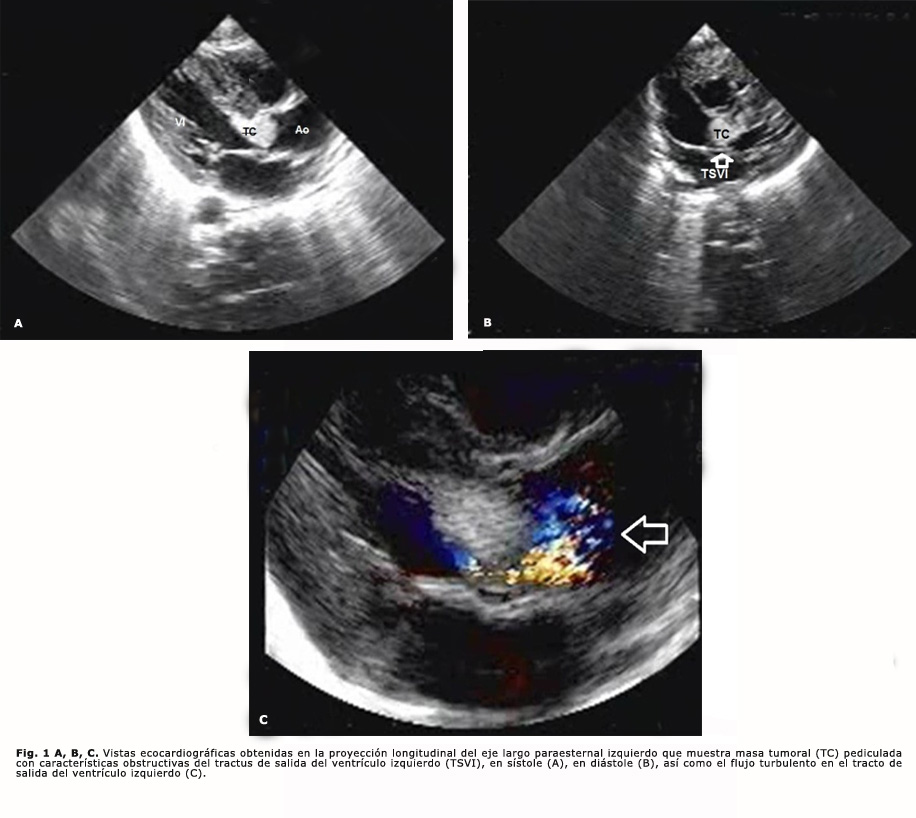
La evaluación ecocardiográfica reveló masa tumoral pediculada con características obstructivas del tractus de salida del ventrículo izquierdo. El doppler continuo reveló gradiente sistólico en aorta ascendente de 76 mmHg; y el doppler color, un flujo sistólico turbulento en aorta ascendente originado a partir del componente obstructivo de la masa tumoral descrita (Fig. 1), regurgitación aórtica mínima asociada, comunicación interauricular (CIA), fosa oval fenestrada de 3,9 mm e hipertrofia biventricular.
El electrocardiograma (ECG) arrojó ritmo sinusal eje QRS derecho, sin signos de isquemia miocárdica; el telecardiograma, cardiomegalia biventricular, tronco de la arteria pulmonar rectificado y flujo pulmonar normal. Se realizaron otros estudios como la tomografía axial computarizada (TAC) de cráneo, ultrasonido renal y fondo de ojo, en los cuales no se evidenciaron otras alteraciones como tuberosidades corticales, nódulos subependimarios, angiomiolipomas renales, hamartomas retinianos, linfangiomatosis pulmonar o lesiones cutáneas.
El paciente se mantuvo asintomático y ecocardiográficamente sin variación, con gradientes sistólicos alrededor de 80 mmHg, por lo que se le indica tratamiento quirúrgico a los 2 meses de edad. Se empleó circulación extracorpórea (CEC), hipotermia ligera y 30 minutos de parada cardíaca con pinzamiento aórtico y cardioplejía cristaloide fría. Se realizó aortotomía oblicua y exéresis de tumor de base ancha, de aspecto nacarado y superficie lisa, que protruye a través de anillo aórtico y comprime seno derecho y parte del izquierdo. Salió espontáneamente de CEC, sin gradiente patológico en tracto de salida del ventrículo izquierdo (TSVI).
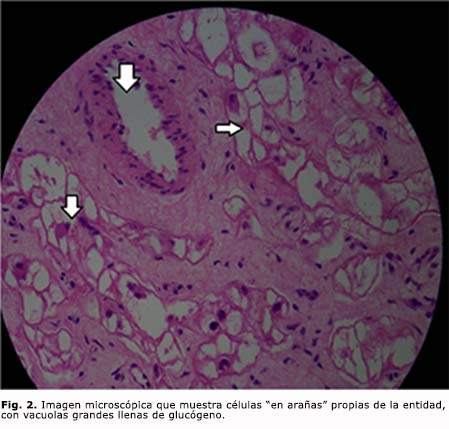
El estudio anatomopatológico describe células vacuoladas con núcleos centrales (células “en arañas”) (Fig. 2). El curso posoperatorio fue sin complicaciones. El paciente evoluciona sin residuos ni secuelas a los 12 meses de seguimiento.
DISCUSIÓN
Como se hizo alusión, la mayoría de los tumores cardíacos primarios en edades pediátricas son rabdomiomas, y en cerca del 80 % se encuentra asociada una esclerosis tuberosa. Se caracterizan por ser circunscritos, lobulados, blanquecinos o grisáceos, habitualmente múltiples hasta en el 90 % de los casos, y generalmente se encuentran en el espesor del músculo cardiaco, que pueden afectar cualquier punto, con predominio de los ventrículos, y de estos, mayormente en el izquierdo.6
Su diagnóstico puede realizarse desde la etapa prenatal, aunque mayoritariamente sucede durante el período neonatal.1 El diagnóstico prenatal se ha realizado desde las 20 semanas de gestación, aunque suele ser más usual en el tercer trimestre del embarazo, y se sospecha ante la presencia de hidrops no inmunológicos, arritmias fetales, crecimiento intrauterino retardado (CIUR) o antecedentes familiares de esclerosis tuberosa.2 En el estudio realizado por Chao y otros, se constató que el tamaño del tumor (> 20 mm), la presencia de arritmia fetal e hidrops, eran fuertes predictores de una mala evolución en el período neonatal.7 En este paciente se realizaron ecocardiografías en los 3 trimestres de la gestación, pero no se visualizó la tumoración intracardiaca. El diagnóstico se hizo en la primera semana de vida, con características descritas en la literatura, pero con la diferencia de ser única, situada en el espesor del músculo cardiaco del TSVI, que protruía a través del anillo aórtico.
En la primera etapa de la vida los signos asociados a la presencia de este tumor se caracterizan por dificultad respiratoria, signos de bajo gasto o de insuficiencia cardíaca, y puede comportarse clínicamente de manera similar a la hipoplasia de cavidades izquierdas.1 Las arritmias son poco frecuentes,8 y se han visto asociadas al cuadro clínico en el 6 % de los pacientes,9 aunque Yadava refiere en su estudio una incidencia ente 16-47 % de los casos.10 En otras ocasiones pasa clínicamente inadvertido, y es un hallazgo en el examen físico la presencia de un soplo secundario a la obstrucción al paso del flujo sanguíneo, como se presentó en este paciente.
La evolución clínica dependerá del número, posición y tamaño del tumor. Puede causar obstrucción marcada al paso del flujo sanguíneo por la cavidad afectada, así como la compresión del sistema de conducción que puede llevar a serios problemas de arritmia. Existe también la posibilidad de embolización de algún fragmento del tumor, o de trombos adheridos a este.1
Entre los complementarios, el ECG, aunque no es específico, puede mostrar la desviación del eje axial a la izquierda, el crecimiento de cavidades, las alteraciones del segmento ST u otros signos de isquemia miocárdica, así como alteraciones del ritmo cardíaco, como son, las taquiarritmias y los bloqueos auriculoventriculares, que pueden ser responsables de episodios de muerte súbita en estos pacientes,1,3 como es el caso reportado por Neri y otros, de muerte súbita en un lactante de 11 meses con diagnóstico de rabdomioma.11
El telecardiograma ofrece pocos datos en muchas ocasiones, principalmente cuando se trata de tumores intracavitarios pequeños. Sin embargo, en otros casos se puede encontrar cardiomegalia y edema pulmonar en recién nacidos y lactantes clínicamente sintomáticos.1
El ecocardiograma bidimensional, la TAC y la resonancia magnética son los principales medios diagnósticos no invasivos para el estudio de los tumores cardíacos. De ellos, el ecocardiograma se ha convertido en una herramienta imprescindible para el diagnóstico de esta enfermedad, porque permite la detección precoz desde la etapa prenatal.1,3
El estudio histológico de la pieza es lo que confirma el diagnóstico.1 Microscópicamente este tumor muestra células “en arañas”, propias de la entidad, con vacuolas grandes llenas de glucógeno. Muchos autores no lo consideran como un tumor realmente, sino como un hamartoma de la fibra muscular estriada.2
El diagnóstico de este paciente se realizó con la ecocardiografía, y se corroboró con el estudio anatomopatológico; se descartaron otras alteraciones que frecuentemente se asocian en el caso de la esclerosis tuberosa.
En la evolución natural del rabdomioma se ha constatado en cerca del 80 % la involución de la masa tumoral durante los primeros años de vida. Después del nacimiento las células del rabdomioma pierden la capacidad de dividirse a través de la mitosis, y comienza su regresión.1,2 Yadava refiere una mortalidad de 5-17 % de los casos operados, con buenos resultados a largo plazo en los restantes casos.10
Este paciente se mantuvo en sus primeros 2 meses de vida asintomático, sin embargo no hubo regresión del tamaño de la tumoración, se mantuvieron gradientes sistólicos a nivel del TSVI altos, y se acompañó de insuficiencia valvular aórtica, lo cual constituye un factor de riesgo importante para provocar arritmias y muerte súbita, por lo cual se decidió tratamiento quirúrgico.
El pronóstico de los niños con diagnóstico de rabdomioma ha mejorado significativamente en las últimas décadas. Se plantea una supervivencia en series estudiadas de hasta 80 % de los casos;12 sin embargo, no se debe olvidar que un buen porcentaje de estos pacientes tiene el diagnóstico de esclerosis tuberosa, entidad también conocida como síndrome de Bourneville, con herencia autosómica dominante y heterogeneidad genética, que empobrece, en muchos de ellos, la calidad de vida futura. Este síndrome genético se caracteriza por desarrollo de lesiones hamartomatosas en múltiples órganos, con predominio en el cerebro, con tuberosidades corticales y nódulos subependimarios. Puede haber angiomiolipomas renales, hamartomas retinianos, linfangiomatosis pulmonar y lesiones cutáneas.
REFERENCIAS BIBLIOGRÁFICAS
1. Moran A, Gerald M. Cardiactumors. In: Allen HD, Driscoll DJ, Shaddy RE, Feltes TF, editors. Moss And Adams Heart Disease in Infants, children and adolescents: Including the Fetus and Young Adults. Philadelphia: Lippincott Williams & Wilkins; 2008. p. 1479-95.
2. Uzun O, Wilson D, Vujanic G, Parson J. Cardiac tumours in children. Orphanet Journal of Rare Diseases. 2007;2(11):44-56.
3. Kohut J, Krzystolik-ładzińska J, Szydłowski L, Smoleńska-petelenz J, Giec-fuglewicz G, Pająk J. The diagnosis, clinical course and follow-up of children with cardiac tumours -a single-centre experience. Kardiologia Polska. 2010;68:304-9.
4. Chávez IM, Castellanos LM, Hernández AB, Faustro AA, Orellana JE, Marroquín SR. Rabdomioma gigante intracardíaco en la etapa neonatal. Reporte de un caso. Arch Cardiol Méx. 2004;74(1):49-52.
5. Pruksanusak N, Suntharasaj T, Suwanrath C, Phukaoloum M. Fetal Cardiac Rhabdomyoma with Hydrops Fetalis. JUM. 2012;31(11):1821-4.
6. López JÁC, Jiménez YG, Gutiérrez LY, Arteaga SJ, Sánchez AM, Cardeña JO, et al. Rabdomioma cardiaco tratado quirúrgicamente con éxito y revisión de la literatura. Arch Cardiol Méx. 2005;75(3):22-4.
7. Chao AS, Chao A, Wang HT, Chang CY, Hsieh CC, Lien R, et al. Outcome of antenatally diagnosed cardiac rhabdomyoma case series and a meta-analysis. Ultrasound in Obstetrics & Gynecology. 2008;31(3):289-95.
8. De Rosa G, Carolis M, Pardeo M, Bersani I. Neonatal emergencies associated with cardiac rhabdomyomas: an 8-year experience. Fetal Diagn. 2011;29(2):169-77.
9. Khatare P, Muthuswamy K, Sadasivan J, Calumbar N. Incessant ventricular tachycardia due to multiple cardiac rhabdomyomas in an infant with Tuberous Sclerosis. Indian Heart J. Jan 2013;65(1):111-3.
10. Yadava OP. Cardiac tumours in infancy. Indian Heart J. 2012;64(5):492-6.
11. Neri M, Di Donato S, Maglietta R, Pomara C. Sudden death as presenting symptom caused by cardiac primary multicentric left ventricle rhabdomyoma, in an 11-month-old baby. An immunohistochemical study. Diagn Pathol. 2012;7:169.
12. Bédard E, Becker AE, AGatzoulis M. Cardiac Tumours. In: Kennedy JF, Suite B, Cardiology P, Livingstone C, editors. Paediatric Cardiology. Philadelphia: Elsevier; 2010. p. 1055-65.
Recibido: 15 de mayo de 2014.
Aprobado: 23 de mayo de 2014.
Mariela Céspedes Almira. Cardiocentro Pediátrico “William Soler”. San Francisco y Perla, reparto Altahabana, municipio Boyeros. La Habana, Cuba. Correo electrónico: marielacespedes@infomed.sld.cu


{kind=link}