










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Pediatría
versión impresa ISSN 0034-7531versión On-line ISSN 1561-3119
Rev Cubana Pediatr vol.87 no.3 Ciudad de la Habana jul.-set. 2015
ARTÍCULO DE REVISIÓN
El electroencefalograma en el síndrome de West y otras entidades clínicas relacionadas
Electroencephalogram in the West syndrome and other related clinical entities
Dr.Cs. Desiderio Rafael Pozo Lauzán, Dra.C. Albia Josefina Pozo Alonso, Dr. José Manuel Sayú Stewart
Hospital Pediátrico Universitario “William Soler’’. La Habana, Cuba.
RESUMEN
Se realiza una revisión de las características electroencefalográficas de los espasmos infantiles, espasmos epilépticos o síndrome de West, y otras entidades relacionadas con este. Se enfatiza en los patrones más frecuentes, fundamentalmente en los 2 tipos de hipsarritmia: clásica y periódica o fragmentada, observados en el síndrome anteriormente mencionado. Se comenta en relación con el trazado de suppression-burst o paroxismos-supresión, y su correlación con los síndromes de Ohtahara, y Aicardi y Goutières, descritos respectivamente en 1976 y 1978. Se aclara que estos 2 patrones no son exclusivos de estos síndromes, y pueden ser observados en otras entidades en el neonato, como la encefalopatía anóxica isquémica, la meningitis neonatal bacteriana y trastornos metabólicos, entre otros.
Palabras clave: síndrome de West, espasmos infantiles, espasmos epilépticos, hipsarritmia.
ABSTRACT
A review was made on the encephalographic characteristics of infantile spasms, epileptic spasms, or West syndrome, and other related entities. Emphasis was made on the most frequent patterns, mainly the two types of hypsarrhythmia, classical and periodic, and fragmented, which are observed in this syndrome. Likewise, comments were made on the suppression-burst or burst-suppression tracing and its correlation with Ohtahara, and Aicardi and Goutières syndromes that were described in 1976 and 1978, respectively. It was clarified that these two patterns are not exclusive of these syndromes and may be also observed in other illnesses affecting the neonates such as anoxic-ischemic encephalopathy, neonatal bacterial meningitis and metabolic disorders, among others.
Keywords: West syndrome, infantile spasms, epileptic spasms, hypsarrhythmia.
INTRODUCCIÓN
Los espasmos infantiles constituyen un grupo de crisis de naturaleza epiléptica que se caracterizan por una contracción brusca —más frecuente bilateral y simétrica— de los músculos del cuello, tronco y miembros. Se acompañan de una breve afectación de la conciencia. Pueden ser en flexión, extensión o mixtos. Usualmente se presentan en series, grupos o salvas. A veces ocurren espasmos aislados. Las series o salvas se manifiestan varias veces al día, y son características al despertar o antes de dormir. Son raros durante el sueño. Es posible observar manifestaciones autonómicas asociadas a la crisis: rubicundez, dilatación pupilar y sudación. Numerosos niños muestran retardo del neurodesarrollo, aunque en algunos pacientes no de detecta previo al comienzo de las crisis. La etapa de comienzo, en la mayoría, ocurre entre los 3 y 6 meses de edad, y un segundo pico entre los 6 y 8 meses. Es menos frecuente la aparición de las crisis después del año de edad. Los espasmos se acompañan a veces de risa y una expresión de susto. Pueden ser precedidos o acompañados por crisis focales, atónicas o tónicas.
La triada de espasmos infantiles, retardo del neurodesarrollo y el patrón electroencefalográfico de hipsarritmia es conocida como síndrome de West.1
El objetivo de este trabajo es realizar una revisión de las principales características electroencefalográficas del síndrome de West y otras entidades relacionadas con este.
DESARROLLO
El patrón electroencefalográfico (EEG) de hipsarritmia es el más común observado en las crisis epilépticas, conocidas como espasmos infantiles, espasmos epilépticos o síndrome de West. Fue descrito por Gibbs y Gibbs en 19522 en un niño despierto. Se caracteriza por un aspecto desorganizado, caótico, casi continuo. Se observan ritmos lentos polimorfos de puntas, y punta-onda lenta de gran amplitud. En ocasiones, los grafoelementos son bilaterales y sincrónicos; y otras veces, se localizan en diferentes focos. Las puntas varían de un momento a otro en duración y localización. En ocasiones parecen ser focales, y pocos segundos después provienen de otro o de múltiples focos. Existe una correlación escasa entre áreas homólogas de ambos hemisferios.
El patrón referido con anterioridad se denomina hipsarritmia clásica o típica, y es característico en vigilia (Fig. 1).
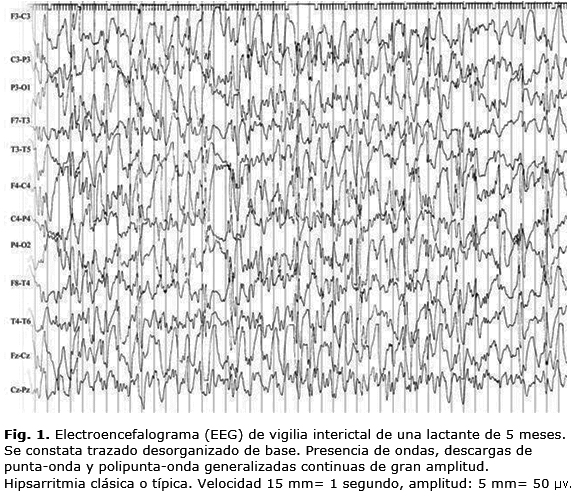
La edad de comienzo de presentación del trazado de hipsarritmia es similar al de los espasmos infantiles. Se observa desde el período neonatal hasta los 2 o 3 años de edad, aunque esto puede ser variable.3
El EEG, fundamentalmente durante la etapa 2 de sueño lento, muestra un aumento de las puntas y polipuntas, y una tendencia a la sincronización, con una fragmentación de la hipsarritmia observada en la vigilia. Este patrón se denomina hipsarritmia periódica o fragmentada4 (Fig. 2). Es posible observarlo en el sueño lento, aunque no esté presente la hipsarritmia típica o clásica durante la vigilia.

El trazado de hipsarritmia puede preceder la aparición de los espasmos infantiles o epilépticos, o puede observarse posteriormente. A juicio de los autores de este trabajo, a través de los años, en la literatura médica mundial han existido ambigüedades en relación con la interpretación de la definición de este patrón EEG, incluidas sus 2 formas: típica, y periódica o fragmentada.
En el servicio de Neuropediatría del Hospital “William Soler”, desde hace años, en estos pacientes, se obtiene un registro de vigilia y además de sueño espontáneo, para poder observar en todos los niños específicamente la etapa 2 del sueño lento, y constatar el patrón de hipsarritmia periódica o fragmentada.
En 1956 Lesse, Hoeffer y Austin5 describieron en 11 pacientes con encefalopatías de diferentes etiologías, un patrón EEG que denominaron oleadas periódicas sincrónicas de ondas lentas y descargas de punta-onda de gran amplitud, alternando con períodos de supresión de la actividad eléctrica cerebral entre los intervalos de las descargas. En 1960, Harris y Tizard6 denominaron trazado paroxístico a un patrón EEG similar al referido anteriormente.
En 1970 Rose y Lombroso7 se refirieron a un trazado EEG con las características de oleadas generalizadas de descargas polimorfas, que alternaban con períodos de supresión de la actividad eléctrica entre estas, y lo denominaron pre-hipsarritmia, debido a que este patrón, en ocasiones, precede al de hipsarritmia fragmentada o periódica.
En la década del 70 Dreyfus-Brisac,8 en el Hospital de Maternidad Port Royal, de París, comenzó a utilizar la terminología de trazado paroxístico, referida con anterioridad por Harris y Tizard,6 y lo asoció a las entidades neonatales siguientes: encefalopatía anóxica isquémica, meningitis bacteriana, trastornos metabólicos como la hiperglicinemia no cetósica, la acidemia metilmalónica y la acidemia propiónica, entre otros.
Este patrón EEG había sido observado por Pozo9 desde el año 1971 en diferentes trastornos en el neonato: meningitis bacteriana, encefalopatía hipóxica-isquémica y espasmos infantiles o epilépticos en lactantes en edades tempranas. Estos pacientes se clasificaban como síndrome de West precoz. El patrón denominado paroxístico fue descrito como suppression-burst en 1977 por Werner.10
Posteriormente, Ohtahara y otros, en Japón,11 describieron en 1976 la encefalopatía epiléptica infantil temprana. Le confirieron valor diagnóstico al denominado trazado de EEG de paroxismos-supresión, por lo que también se conoce como síndrome de Ohtahara.
Las crisis principales referidas consisten en espasmos tónicos que ocurren en series o salvas, o en forma esporádica. Se observan en vigilia o durante el sueño. También pueden constatarse crisis parciales motoras erráticas, crisis motoras unilaterales y en báscula.
En 1978 Aicardi y Goutières,12 en Francia, describieron lo que denominaron encefalopatía mioclónica precoz. Los autores también le otorgaron valor diagnóstico al patrón EEG referido anteriormente. De acuerdo con estos autores,12 se caracteriza por mioclonías fragmentarias, y se acompañan de crisis parciales motoras, espasmos tónicos y otras crisis.
Los autores de este trabajo opinan que los síndromes descritos por Ohtahara y otros,11 Aicardi y Goutières12 y el síndrome de West, constituyen una única entidad nosológica, que tiene diferentes formas de expresión clínica y EEG en función de la maduración cerebral.
Es posible que algunos casos descritos por Ohtahara y otros,11 y posteriormente por Aicardi y Goutières,12 correspondan a espasmos infantiles (síndrome de West), que se manifiesten en el neonato de forma precoz. Se hace la pregunta de qué diferencia existe entre los espasmos tónicos, incluso, agrupados en series o salvas referidos en ambos síndromes, y los clásicos espasmos infantiles, epilépticos o síndrome de West.
Se han descrito trazados considerados como hipsarritmia atípica o modificada (Hrachovy y otros),4 aunque esta terminología ha sido polémica a través de los años:
- Patrón de paroxismos-supresión (Fig. 3). Esta variante de hipsarritmia, como se expuso con anterioridad, puede observarse en otras entidades clínicas del neonato.
- Hipsarritmia asimétrica, e incluso, unilateral.
- Asociación de la hipsarritmia con un foco de descargas fundamentalmente temporales.
- Conservación parcial de la actividad de base con uno o varios focos individuales, y de oleadas de puntas-ondas lentas generalizadas.
- Existe una variante de hipsarritmia denominada lenta, que se caracteriza por un predominio de esa actividad con pocas puntas.
- La variante rápida se caracteriza por el predominio de esta frecuencia con pocas puntas.
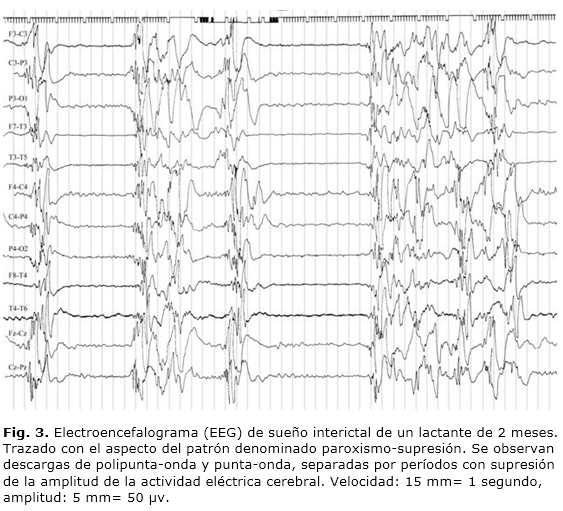
Recientemente, han aparecido nuevas referencias en la literatura médica internacional en relación con el trazado de paroxismos-supresión que a continuación se comentan:
- En neonatos cuyos períodos de supresión entre las descargas, son muy prolongados. Estos se asociaron a daño severo de la sustancia blanca cerebral.13
- En recién nacidos con el diagnóstico de asfixia perinatal el trazado de paroxismos-supresión constituyó un marcador de daño cerebral reciente.14
- Se confirmó15 que este patrón de paroxismos-supresión se asocia a pobre pronóstico.
- Se reportó16 mal pronóstico en un grupo de pacientes con el diagnóstico de coma posanóxico, en los que el EEG se caracterizaba por el patrón de paroxismos-supresión.
- Se sugirió en un recién nacido con la presencia del trazado de paroxismos-supresión, y que tuvo una respuesta favorable a la vigabatrina, que podría tratarse de un síndrome de West de comienzo precoz,17 como se había comentado con anterioridad.
Hasta ahora se han expuesto patrones interictales (fuera de las crisis), aunque también se ofrecen las características EEG de los espasmos infantiles o epilépticos en el momento de las crisis.
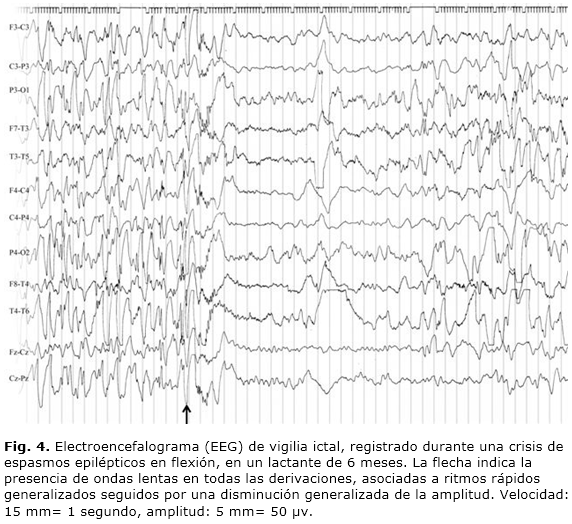
En relación con los patrones ictales del EEG, en los espasmos epilépticos no se ha insistido tanto en la literatura médica mundial como en los interictales. No obstante, se han referido diversos tipos de actividades ictales3,18-20 que se exponen a continuación: la más frecuente constatada la constituye la presencia de ondas lentas generalizadas de elevada amplitud, sobreimpuestas por una actividad rápida de bajo voltaje. Puede ser seguida por una disminución o decremento difuso del voltaje (Fig. 4), ritmos rápidos generalizados, complejos generalizados de puntas lentas y punta-onda lenta.
CONSIDERACIONES FINALES
En los espasmos infantiles, epilépticos o síndrome de West, existe un patrón EEG interictal denominado hipsarritmia, que es el más frecuente en sus 2 modalidades: típica (durante la vigilia), y la fragmentada (durante el sueño lento), y se ha considerado característico a través de los años, a pesar que este término tiene algunas ambigüedades en su definición. El patrón ictal más característico en esta entidad, es la presencia de ondas lentas de gran amplitud, sobreimpuestas por una actividad rápida de escasa amplitud.
Sin embargo, el patrón de paroxismos-supresión, que se ha definido como criterio diagnóstico específico en la encefalopatía epiléptica infantil temprana (síndrome de Ohtahara) y la encefalopatía mioclónica neonatal de Aicardi y Goutières, puede observarse en otras entidades que no necesariamente corresponden a esos 2 síndromes epilépticos mencionados previamente. La fisiopatología de este patrón EEG no está completamente dilucidada.
Se recomienda seguir revisando las definiciones de los diferentes patrones EEG referidos en este trabajo, con el objetivo de unificar criterios, y evitar errores y ambigüedades en su interpretación. Es evidente que esto contribuirá, sin lugar a dudas, a lograr un diagnóstico más confiable de estas entidades.
REFERENCIAS BIBLIOGRÁFICAS
1. Hrachovy RA, Frost J. Infantile Spasms. In: Dam l, editor. Comprehensive epileptology. New York: Raven Press; 1990. p. 113-21.
2. Gibbs FA, Gibbs EL. Atlas of Electroencephalography. Vol 2. Epilepsy. Cambridge: Adison Wesley; 1952. p. 16-1.
3. Pozo Lauzán D, Pozo Alonso A. Atlas de electroencefalografía en el niño. La Habana: Editorial Ciencias Médicas; 2013. p. 227-35.
4. Hrachovy RA, Frost JD, Kellaway P. Hypsarrhythmia: variations on the theme. Epilepsia. 1984;25:17-25.
5. Lesse PE, Hoeffer A, Austin JH. Difusse encephalopathy associated with periodic synchronous abnormal discharges in the electroencephalogram. Trans Amer Neurol Ass. 1956;81:74-82.
6. Harris R, Tizard JPM. The electroencephalogram in neonatal convulsions. Pediatrics. 1960;57:501-20.
7. Rose AL, Lombroso CT. Neonatal Seizure State. Pediatrics. 1970;45:404-25.
8. Dreyfus-Brisac C. Neonatal Electroencephalography. In: Scarpelli E, Cosmi EV, editor. Reviews in Perinatal Medicine. Vol 3. New York: Raven Press; 1979. p. 15.
9. Pozo D. Neonatal Convulsions: a clinical and electroencephalographic study [tesis de doctorado]. Universidad de Karolinska, Praga; 1981. p. 17.
10. Werner SS, Stocjard JE, Rickford RG. Atlas of Neonatal Electroencephalograpjy. New York: Raven Press; 1977. p. 28-9.
11. Ohtahara S, Ishida T, Oka E. On the specific age-dependent epileptic syndrome. The early-infantile epileptic encephalopathy with suppression-burst. No to Hattasu. 1976;8:270-80.
12. Aicardi J, Goutières E. Encephalopathie myoclonique Neonatal. Rev EEG Neurophysiology. 1978;8:99-101.
13. Liu YF, Tong YM, Zhou CL, Zhang DD, Piao MH. Relationship between degree of white matter damage and EEG changes in premature infants early after birth. Zhongguo Dang Dai er Kezazhi. 2013;15:321-6.
14. Iyer KK, Roberts JA, Metsaranta M, Finnigan S, Breakspear M, Vannatalo S. Novel features of early burst-suppression predict outcome after birth asphyxia. Ann Clin Transl Neurology. 2014;1:209-14.
15. Soholm H, Kjaer TW, Kjaergaard J, Cronberg T, Bro-Jeppesen J, Lipert FK, et al. Prognostic value of electroencephalography (EEG) after out-of hospital cardiac arrest in successfully resuscitation patients used in daily clinical practice. Resuscitation. 2014;85:1580-5.
16. Hofineijer J, Tjepkema-cloostermans MC, Vanputter MJ. Burst-suppression with identical bursts. A distinct EEG pattern with poor outcome in postanoxic coma. Clin Neurophysiol. 2014;125:947-54.
17. Korff CM, Vulliemoz S, Picard F, Fluss J. Ohtahara Syndrome or early onset of West Syndrome? A case with overlapping features and favorable response to Vigabatrin. Eur J Paediatr Neurol. 2012;16:753-7.
18. Lombroso CT. A prospective study of infantile spasms: clinical and therapeutic correlations. Epilepsia. 1983;24:135-58.
19. Fusco L, Vigevano F. Ictal Clinical electroencephalographic findings of spasms in West syndrome. Epilepsia. 1993;34:671-8.
20. Nash K, Sullivan J. Myoclonic seizures and infantile spasms. En: Swaiman KF, Ashwal S, Ferriero Schor NF, eds. Pediatric Neurology. 5th ed. Philadelphia: Saunders, Elsevier; 2012. p. 774-89.
Recibido: 19 de marzo de 2015.
Aprobado: 1ro. de abril de 2015.
Desiderio Rafael Pozo Lauzán. Hospital Pediátrico Universitario “William Soler”. Calle 100 y Perla, Reparto Altahabana, municipio Boyeros. La Habana, Cuba.
Correo electrónico: pozo@infomed.sld.cu

