










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Pediatría
versión On-line ISSN 1561-3119
Rev Cubana Pediatr vol.88 no.4 Ciudad de la Habana oct.-dic. 2016
Rev Cubana Pediatr. 2016;88(4)
PRESENTACIÓN DE CASO
Severos trastornos neurológicos y malformaciones en una niña con monosomía del cromosoma 21
Severe neurological disorders and malformations seen in a girl with chromosome 21 monosomy
Luis Alberto Méndez-Rosado,I Gisela Noche González,II Tatiana Zaldívar Vaillant,III Luanda Maceiras Rosales,I Yomisleydy Bravo ÁlvarezI
ICentro Nacional de Genética Médica. La Habana, Cuba.
IICentro Provincial de Genética Médica. Villa Clara, Cuba
IIIHospital Nacional de Neurología. La Habana, Cuba.
RESUMEN
Introducción: la monosomía del cromosoma 21, generalmente es incompatible con la vida, aunque se reportan casos en los que esta aparece en forma de mosaico, y constituye un hallazgo inusual en los estudios genéticos realizados en Pediatría.
Presentación del caso: niña de 7 años de edad, que fue remitida a la consulta de Asesoramiento Genético por presentar malformaciones congénitas severas y rasgos dismórficos, asociado a un retardo del neurodesarrollo. Al nacer se diagnosticó una comunicación interauricular, que requirió cirugía. Se le realizó estudio por técnicas de citogenética convencional y se obtuvo como resultado una inusual monosomía del cromosoma 21. El estudio de técnica de citogenética molecular detectó una inserción de la zona crítica del 21 en la región subtelomérica del 6p.
Conclusiones: el correcto examen clínico de la paciente, unido a los métodos moleculares empleados, permite establecer una hipótesis del diagnóstico de este caso tan inusual.
Palabras clave: trastornos neurológicos; malformaciones; monosomía 21; FISH.
ABSTRACT
Introduction: chromosome 21 monosomy is generally life-threatening, although cases are reported in which it appears in mosaic structure and represents an unusual finding in genetic studies carried out in pediatrics.
Case presentation: a seven years-old girl was referred to the genetic counseling service because she presented with severe congenital malformations and dimorphic traits associated to retardation in her neurodevelopment. When she was born, she was diagnosed with interatrial communication requiring surgery. This patient was studied with conventional cytogenetic techniques and the result was an unusual chromosome 21 monosomy. The molecular cytogenetic study detected insertion of the critical area of chromosome 21 into the subtelomeric region of chromosome 6p.
Conclusions: adequate clinical examination of the patient together with the molecular testing methods allows setting a diagnostic hypothesis of this unusual case.
Keywords: neurological disorders; malformations; monosomy 21; FISH.
INTRODUCCIÓN
Las aberraciones cromosómicas de tipo estructural implican cambios en la secuencia lineal de los genes sobre los cromosomas, que pudiesen provocar pérdidas, ganancias o reordenamiento de sus secciones particulares. Se presentan en la población con una frecuencia de 1 en 500 individuos, y en ocasiones, afectan a más de un cromosoma. Las inserciones cromosómicas son eventos raros. Con los métodos de la citogenética clásica Van Hemel and Eussen estimaron su frecuencia en 1 en 80 000, pero actualmente se conoce que, aplicando técnicas de citogenética molecular, su frecuencia es mucho mayor.1,2
La monosomía del cromosoma 21, generalmente es incompatible con la vida, aunque se reportan casos en los que esta aparece en forma de mosaico, y otros, en que al ser evaluados por las técnicas moleculares, resultan en monosomía parcial del cromosoma 21, que pueden ser heredados de rearreglos cromosómicos balanceados en los padres, o tener un origen de novo.3,4
Una inusual monosomía del cromosoma 21 con sus rasgos fenotípicos es presentada y se establece la posible correlación cariotipo-fenotipo.
PRESENTACIÓN DEL CASO
Se remite una niña de 7 años con múltiples rasgos dismórficos, severa cifoescoliosis de la región torácico-lumbar y discapacidad intelectual, a la consulta de Asesoramiento Genético. Pesó al nacer 2 000 g y su talla fue de 48 cm. Se diagnostica en los primeros días de vida una cardiopatía congénita (CIA), cerrada por cateterismo a los 4 años. Presenta infecciones respiratorias frecuentes.
Examen físico: baja talla para la edad (106 cm). Cráneo y cara: macro cráneo (36,4 cm), con frente muy amplia, abombada, cabello de implantación muy alto -a nivel de la frente-, marcado hipertelorismo, puente nasal deprimido, pestañas muy curvas, asimetría de iris (coloboma), filtro amplio, labio superior extremadamente fino, narinas evertidas, apiñamiento de dientes en ambas arcadas, ausencia congénita de incisivos laterales superiores, paladar alto y ojival, orejas grandes displásicas de muy baja implantación, cara alargada y mentón prominente. El cuello era corto, con limitación a los movimientos; y el tórax deformado, de forma cilíndrica, con pectus carinatum, sin hallazgos de interés en el abdomen a la inspección, ni visceromegalia.
Las extremidades y columna vertebral presentaban miembros superiores con deformidad de ambos codos, que se encuentran en posición en abducción con hiperlaxitud a ese nivel, con manos largas de dedos largos y finos. Los miembros inferiores presentaban atrofia muscular en ambos, y flaccidez a ese nivel. En la columna vertebral se observa cifoescoliosis dorso-lumbar grave, con presencia de giba izquierda, los hombros son asimétricos.
En relación con el sistema nervioso central, se observa hipotrofia en miembros inferiores, debilidad muscular del tren inferior, hiporreflexia osteotendinosa, fuerza muscular disminuida sin control del tronco, severo trastorno para la marcha, discapacidad intelectual severa, así como retardo del lenguaje.
Estudios complementarios realizados:
- Ecosonograma transfontanelar a los 15 días de edad: asimetría de ventrículos laterales con ventriculomegalia izquierda. Quiste subependimario derecho.
- Potenciales evocados visuales: percepción del estímulo pero no permitió concluir sobre la calidad de la visión.
- Potenciales evocados del tallo cerebral: ausencia de respuesta del lado derecho compatible con pérdida auditiva completa, con hipoacusia mixta neurosensorial severa del lado izquierdo, distrofia y adelgazamiento del cuerpo calloso.
- Estudios con resonancia magnética nuclear (RMN):
- RMN cerebral (8 meses de edad). Dilatación de tipo triventricular con presencia de prolongaciones temporales de los ventrículos laterales, distrofia y adelgazamiento del cuerpo calloso, asimetría de cuernos occipitales, dilatación de la cisterna quiasmática y prepontina con disminución de la cisterna cuadrigémina.
- RMN cerebral (2 años de edad). Discreta hidrocefalia, cambios a nivel del cuerpo calloso en relación con probable hipoplasia.
- RMN de columna cervical, dorsal y lumbar (2 1/2 años de edad). Acentuada rotoescoliosis dorsal de convexidad derecha, el canal raquídeo de amplitud conservada, la médula espinal se extiende hasta la vértebra lumbar 1 (L1) sin evidencia de alteraciones.
- Rayos x de caderas y de ambos codos: estructuras óseas evaluadas de morfologías y densidad radiológica conservada. Espacios articulares conservados, sin alteraciones de partes blandas.
- Rayos x de columna vertebral: se evidencia curva toracolumbar (TL) T5 a L2 con convexidad derecha desbalanceada.
- Estudios cromosómicos convencionales en sangre periférica:
- La niña presentó monosomía del cromosoma 21 en 50 metafases analizadas a una resolución de 400 bandas (45,XX,-21).
- Es interconsultada con especialista en Genética Clínica, para evaluar la posibilidad de realizar cariotipo a ambos padres y estudio cromosómico a la niña mediante técnica de citogenética molecular (FISH), lo cual se lleva a cabo con los resultados siguientes:
- Cariotipo del padre: 46,XY,inv(9)(p11,q12) en 20 metafases. Cariotipo de la madre: 46,XX en 20 metafases (ambos cromosómicamente normales).
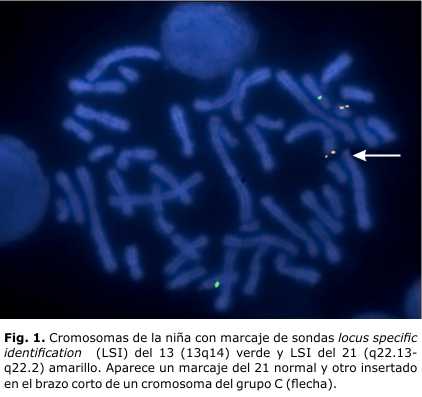
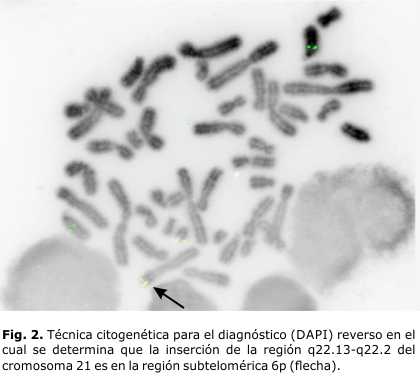
- Estudio cromosómico por FISH a la niña: la zona crítica del 21 está insertada al 6p (Figs. 1 y 2).
- Fórmula cromosómica: 45,XX,-21.ish der(6)ins(6;21)(p25;21q22.13q22.2)(LSI 21 x2)
- Cariotipo del padre: 46,XY,inv(9)(p11,q12) en 20 metafases. Cariotipo de la madre: 46,XX en 20 metafases (ambos cromosómicamente normales).
DISCUSIÓN
El hecho de que esta niña sobreviviera hasta los 7 años con solo un cromosoma 21, se debe a la presencia de la región crítica del cromosoma 21 insertada al 6p, lo cual descartó una monosomía total de ese cromosoma. El cromosoma 21 presenta una baja densidad génica, solo 225 loci codifican proteínas.5 Esta escasez de genes explica que sea el síndrome cromosómico de mayor sobrevivencia, a pesar de su estado trisómico. En esta niña la llamada región crítica, 21q22, está presente en disomía, lo cual sugiere que muchas de las características fenotípicas que muestra no son debidas a la monosomía parcial de este cromosoma. La región 21q22.13q22.2 en triple dosis, está asociada a las principales características fenotípicas del síndrome de Down, por lo cual constituye una zona clave dentro del cromosoma 21.
Las características del cuadro clínico de la niña (anomalías esqueléticas, baja talla, coloboma del iris, discapacidad intelectual, retardo del lenguaje, pérdida de audición, cardiopatías congénitas, entre otras), se corresponden con lo reportado por varios autores en pacientes que presentan deleción 6pter, sobre todo, las subteloméricas.6-12 La deleción de la región subtelomérica del 6p, desde 6p24 hasta 6p25 incluye aproximadamente 5,0 megabases, y es imposible de diagnosticar con el nivel de resolución con que fue analizado este caso por citogenética convencional. Esta deleción ha sido asociada con el síndrome Ritscher-Schinzel (o síndrome cráneo-cerebelo-cardiaco [3C síndrome]).9 Las características clínicas de la paciente sugieren que, debido a la inserción de la región 21q22 en la zona terminal del 6p, se pudo haber producido una deleción subtelomérica o disrupción de genes de esa región, que provocaron este fenotipo. Para un diagnóstico más certero sería necesaria una alta tecnología (micro arreglos de 180 kb), con la que no contamos en este momento en nuestro país.
Las inserciones se consideran un tipo de "translocación no recíproca", que, dependiendo el punto de ruptura donde ocurra la inserción, pueden ser completamente balanceadas, o provocar serios desbalances debido a disrupción de genes codificantes de proteínas con función estructural o genes con función reguladora, deleción o duplicación de genes en el punto de inserción del nuevo fragmento, alteraciones en los CNV (copy number variation por sus siglas en inglés), que tiene que ver con la dosis génica y las interacciones entre genes y los mecanismos de su regulación.13-21 Si tenemos en cuenta que la inserción ha ocurrido en una región rica en genes, como las subteloméricas, es muy posible que esta gran cantidad de afecciones fenotípicas se deban, principalmente, al punto de ruptura en el cromosoma 6, y no estén mayormente relacionadas con la monosomía parcial del cromosoma 21.
Se concluye que el correcto examen clínico de la paciente, unido a los métodos moleculares empleados, ha permitido establecer una hipótesis del diagnóstico de este caso tan inusual.
CONFLICTO DE INTERESES
Los autores declaran no tener conflicto de intereses en la realización del estudio.
REFERENCIAS BIBLIOGRÁFICAS
1. Van Hemel JO, Eussen HJ. Interchromosomal insertions. Identification of five cases and a review. Hum Genet. 2000;107:415-32.
2. Kang SHL, Shaw C, Ou Z, Eng PA, Cooper ML, Pursley AN, et al. Insertional translocation detected using FISH confirmation of array-comparative genomic hybridization (aCGH) results. Am J Med Genet. 2010;152A:1111-26.
3.Martinez-Garcia M, Ainse E, García-Hoyos M, Bustamante A, Cardero R, Ramos-Corrales C, et al. Broadening our understanding by the use of molecular cytogenetic techniques: full monosomy 21. J Assist Reprod Genet. 2011;28(7):106-11.
4. Roberson ED, Wohler ES, Hoover-Fong JE, Lisi E, Stevens EL, Thomas GH, et al. Genomic analysis of partial 21q monosomies with variable phenotypes. Eur J Hum Genet. 2011;19(2):235-8.
5. Hattori M, Fujiyama A, Taylor TD, Watanabe H, Yada T, Park HS, et al. The DNA sequence of human chromosome 21. Nature. 2000;405:311-9.
6. Vernon HJ, Bytyci A, Batista D, Owegi M, Leigh R. 6p25 microdeletion: white matter abnormalities in an adult patient. Am J Med Genet A. 2013;161A(7):1686-9.
7. Piccione M, Antona R, Salzano E, Cavani S, Malacarne M, Morreale Bubella R, et al. Array-CGH and clinical characterization in a patient with subtelomeric 6p deletion without ocular dysgenesis. Am J Med Genet A. 2012 Jan;158A(1):150-4.
8. Martinet D, Filges I, Besuchet N, Morris MA, Gaide AC, Dahoun S, et al. Subtelomeric 6p deletion: clinical and array-CGH characterization in two patients. Am J Med Genet A. 2008;146A(16):2094-102.
9. Descipio C, Schneider L, Young TL, Wasserman N, Yaeger D, Lu F, et al. Subtelomeric deletions of chromosome 6p: molecular and cytogenetic characterization of three new cases with phenotypic overlap with Ritscher-Schinzel (3C) syndrome. Am J Med Genet A. 2005;134A(1):3-11.
10. Le Caignec C, De Mas P, Vincent MC, Boceno M, Bourrouillou G, Rival JM, et al. Subtelomeric 6p deletion: clinical, FISH, and array CGH characterization of two cases. Am J Med Genet A. 2005;132A(2):175-80.
11. Zirn B, Hempel M, Hahn A, Neubauer B, Wagenstaller J, Rivera-Bruguès N, et al. Polyneuropathy, scoliosis, tall stature, and oligodontia represent novel features of the interstitial 6p deletion phenotype. Am J Med Genet A. 2008;15;146A(22):2960-5.
12. Nakane T, Kousuke N, Sonoko H, Yuko K, Sato H, Kubota T, et al. 6p subtelomere deletion with congenital glaucoma, severe mental retardation, and growth impairment. Pediatr Int. 2013;55(3):376-81.
13. Sismani C, Kitsiou-Tzeli S, Ioannides M, Christodoulou C, Anastasiadou V, Stylianidou G, et al. Cryptic genomic imbalances in patients with de novo or familial apparently balanced translocations and abnormal phenotype. Molecular Cytogenetics. 2008;1:1-15.
14. Gribble SM, Prigmore E, Burford DC, Porter KM, Ling NB, Douglas EJ, et al. The complex nature of constitutional de novo apparently balanced translocations in patients presenting with abnormal phenotypes. J Med Genet. 2005;42:8-16.
15. Le Scouarnec S, Gribble SM. Characterising chromosome rearrangements: recent technical advances in molecular cytogenetics. Heredity. 2012;108:75-85.
16. Bache I, Hjorth M, Bugge M, Holstebroe S, Hilden J, Schmidt L, et al. Systematic re-examination of carriers of balanced reciprocal translocations: a strategy to search for candidate regions for common and complex diseases. Europ J Human Genet. 2006;14:410-7.
17. Brady PD, Chiaie BD, Christenhusz G, Dierickx K, Van Den Bogaert K, Menten B, et al. A prospective study of the clinical utility of prenatal chromosomal microarray analysis in fetuses with ultrasound abnormalities and an exploration of a framework for reporting unclassified variants and risk factors. Genetics in Medicine. 2014;16:1-6.
18. Baptista J, Mercer C, Prigmore E, Gribble SM, Carter N, Maloney V, et al. Breakpoint Mapping and Array CGH in Translocations: Comparison of a Phenotypically Normal and an Abnormal Cohort. The American Journal of Human Genetics. 2008;82:927-36.
19. Henrichsen C, Chaignat E, Reymond A. Copy number variants, diseases and gene expression. Human Molecular Genetics. 2009;18:1-8.
20. Mills RY, Walter K, Stewart C, Handsaker RE, Chen K, Alkan C, et al. Mapping copy number variation by population scale genome sequencing. Nature. 2011;470(7332):59-65.
21. Qiao Y, Badduke Ch, Mercier E, Lewis SM, Pavlidis P, Rajcan-Separovic E. miRNA and miRNA target genes in copy number variations occurring in individuals with intellectual disability. BMC Genomics. 2013;14:544-54.
Recibido: 1ro. de diciembre de 2015.
Aprobado: 30 de marzo de 2016.
Luis Alberto Méndez-Rosado. Centro Nacional de Genética Médica. Avenida 31 No. 3 102, esquina a 146, reparto Cubanacán, municipio Playa. La Habana, Cuba.
Correo electrónico: albermen@infomed.sld.cu

