










Meu SciELO
 Serviços customizados
Serviços customizadosServiços Personalizados
Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkRevista Cubana de Pediatría
versão On-line ISSN 1561-3119
Rev Cubana Pediatr vol.89 no.1 Ciudad de la Habana jan.-mar. 2017
PRESENTACIÓN DE CASO
Tumor estromal gastrointestinal
Gastrointestinal stromal tumor
Caridad Verdecia Cañizares,I Ramón Villamil Martínez,II Idalmis Montero Reyes,III Damián Pineda FernándezIV
IServicio de Oncología. Hospital Pediátrico Universitario "William Soler". La Habana, Cuba.
IIServicio de Transplante Hepático. Hospital Pediátrico Universitario "William Soler". La Habana, Cuba.
IIIServicio de Gastroenterología. Hospital Pediátrico Universitario "William Soler". La Habana, Cuba.
IVDepartamento de Anatomía Patológica. Hospital Pediátrico Universitario "William Soler". La Habana, Cuba.
RESUMEN
Introducción: el tumor estromal gastrointestinal es un raro tumor pediátrico que asienta generalmente en estómago en el 50 % de los casos, seguida de su localización en intestino delgado. En el intestino grueso es más rara, al igual que en esófago. Puede ir desde una forma benigna a una forma maligna de la enfermedad. Existe una gran cantidad de factores predisponentes, pero se le concede gran valor a la predisposición hereditaria y a las mutaciones, y también se asocia a la neurofibromatosis tipo 1. Se manifiesta clínicamente con sangrado digestivo oculto, anemia y vómitos. Se describen molestias digestivas y dolor abdominal.
Presentación del caso: se presenta a la comunidad médica el caso de un paciente masculino, de 15 años de edad, mestizo, con un tumor estromal en la pared gástrica posterior al nivel de la curvatura mayor, muy cerca del píloro. El tumor fue resecado quirúrgicamente mediante laparotomía, pero el diagnóstico inicial y toma de biopsia se realizó por vía de cirugía de mínimo acceso. El paciente actualmente está bajo vigilancia estrecha.
Conclusión: existen pocos casos publicados de tumor estromal gastrointestinal en Pediatría y se resalta la importancia de la cirugía en el control de la enfermedad.
Palabras clave: tumor estromal gástrico; Pediatría.
ABSTRACT
Introduction: the gastrointestinal stromal tumor is a rare pediatric tumor that is generally located in the stomach in 50 % of patients, followed by the location in the small intestines. It is even rarer in the large intestine as well as in the esophagus. It may range from a benign to a malignant form of the disease. There is a large number of predisposing factors, but great importance is attached to hereditary predisposition and mutations and is also associated to type 1 neurofibromatosis. Its clinical manifestations are occult digestive bleeding, anemia and vomiting. Digestive upsets and abdominal pain were described.
Case presentation: this is the case of a male, mixed race patient aged 15 years, who presented with a stromal tumor in the posterior gastric wall at the level of major curvature, very close to the pylorus. The tumor was surgically resected through laparotomy, but the initial diagnosis and the biopsy taking was made by way of minimal access surgery. The patient is under close follow-up.
Conclusions: there are few cases that have been published about gastrointestinal stromal tumor in pediatrics and the importance of surgery for the control of this disease is underlined.
Keywords: gastric stromal tumor; Pediatrics.
INTRODUCCIÓN
Existen diferentes tipos de lesiones que se asientan en la pared gástrica, como: el leiomioma, el leiomioma celular, el leiomiosarcoma y el tumor estromal gastrointestinal (GIST). Este último representa de 0,1 a 3 % de todas las neoplasias intestinales, puede abarcar un amplio espectro, desde lesiones benignas hasta tumores altamente agresivos, muchas veces asintomáticos, y su diagnóstico se logra por hallazgos endoscópicos o por estudios imagenológicos; otras veces, se descubren durante una autopsia.1
Se pueden presentar, según su localización, en cualquier zona de la pared gástrica, y pueden presentarse desde formas asintomáticas a casos que se presenten como una urgencia quirúrgica por perforación gástrica o sangrado digestivo.1,2 El diagnóstico de estas lesiones se basa en el estudio microscópico con técnicas de inmunohistoquímica (IHQ).1-3
La endoscopia digestiva con biopsia aporta gran cantidad de datos. Se plantea también el uso de la ultrasonografía endoscópica para su diagnóstico; pues, estudios contrastados del tracto gastrointestinal superior, muestran el defecto de lleno en la pared gástrica, y la tomografía axial computarizada (TAC) permite visualizar el tumor exofítico que se origina en la pared del estómago u otras partes del tubo digestivo, bien delimitado, que puede presentar hemorragia, necrosis o componente quístico. El tratamiento es la resección quirúrgica completa por vía del método laparoscópico o por vía laparotómica.1-3 Existe riesgo de recurrencia del tumor después de la cirugía, por una resección quirúrgica deficiente, o por patrones genéticos encargados de la reaparición de la enfermedad.4
PRESENTACIÓN DEL CASO
Paciente masculino, de 15 años de edad, mestizo, con antecedentes de buena salud hasta el 7 de julio de 2015, fecha en que comenzó con epigastralgia y anorexia ligera. Fue valorado en su área de salud y el estudio detectó anemia de 5,6 g/L, por lo que se le impuso tratamiento con sales de hierro vía oral y vitamina C. Es remitido al Instituto de Hematología e Inmunología, donde se ingresa a inicio de febrero de 2016 y se le estudia. Se descarta hemopatía maligna, es interconsultado con los médicos del servicio de Gastroenterología de nuestro hospital, quienes lo trasladan a su servicio, y continúan el estudio. Detectan sangre oculta en heces fecales y le indican ultrasonido de abdomen, que no muestra afectación en órganos intraabdominales, y deciden realizar endoscopia digestiva alta, en la cual visualizan erosión de la mucosa gástrica y engrosamiento intramural en la pared posterior a nivel de la curvatura mayor de aspecto tumoral, por lo que se toma biopsia de la mucosa gástrica que mostró gastritis crónica. Se discute en equipo multidisciplinario, y se decide realizar laparoscopia con toma de biopsia de la lesión a finales del mismo mes, y el resultado de anatomía patológica resultó ser GIST de bajo grado de malignidad.
Se decide operar el 7 de marzo mediante laparotomía con resección gástrica distal y del píloro, y se hizo gastroyeyunostomía con el proceder de Billrout II. La recuperación posoperatoria fue muy buena, se impuso tratamiento antibiótico con ceftriaxona, amikacina y metronidazol endovenoso. A los 7 días siguientes a la operación, estando con sonda nasogástrica durante las primeras 72 h del posoperatorio, se inició la vía oral con líquidos (al sexto día de operado), con muy buena tolerancia. La resección gástrica distal incluyó el tumor en curvatura mayor, que macroscópicamente medía 3,5 cm × 3 cm × 3 cm, con consistencia fibroelástica, superficie rugosa y color pardo. El tumor no invade la mucosa ni la serosa gástrica (Fig. 1).
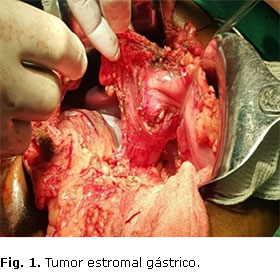
El tumor fue completamente resecado, y no se encontraron otros en los bordes de sección quirúrgica (Fig. 2).
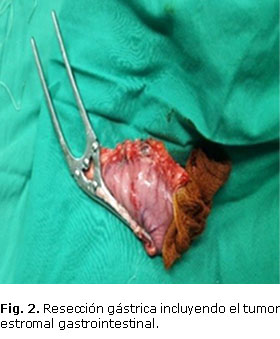
El resultado de la biopsia con IHQ realizada en el Centro Nacional de Referencia de Anatomía Patología del Hospital "Hermanos Amejeiras'' fue: GIST, KI 167 positivo en el 1 % de los núcleos tumorales, CD 117 y CD 34 positivos +++, alfactina negativa y S 100 negativa.
La recuperación posoperatoria fue satisfactoria, y el paciente fue dado de alta a los 15 días de la cirugía totalmente recuperado. Se decidió no iniciar el tratamiento con imatinib (Gleevec) por su toxicidad en edad pediátrica, y realizar el seguimiento por consulta de Gastroenterología y Oncopediatría, inicialmente con una frecuencia mensual durante un año. Después, el seguimiento será trimestral en el segundo año, y semestral el tercer año. A partir de entonces, será con frecuencia anual, hasta completar los cinco años, con el objetivo de detectar cualquier manifestación de recidiva tumoral.
DISCUSIÓN
El GIST es una neoplasia mesenquimal gastrointestinal, que va desde lesiones benignas hasta sarcomas metastásicos, y se origina en el tracto gastrointestinal, omento, mesenterio y retroperitoneo, pero tiene predilección por el estómago y el intestino delgado. El 50-60 % de los casos se asienta en estómago, del 20-30 % en intestino delgado, el 10 % en colon (recto) y 5 % en esófago.1-4
El GIST puede originarse en células intersticial de Cajal, las cuales son similares a marcapasos intermediarios entre el sistema nervioso autónomo gastrointestinal y las células del músculo liso, que regulan la actividad gastrointestinal y el funcionamiento del sistema nervioso autónomo;2,5 pero la enfermedad solo logró distinguirse de otras entidades, como leiomiomas y leiomiosarcoma, con técnicas de IHQ, ya que estas combinan elementos neurales y musculares, y tienen un inmunofenotipo similar en la microscopía electrónica.5 Se plantea que el 85 % de los GIST contienen mutaciones oncogénicas en uno de dos receptores de tirosina cinasa: KIT o PDGFRA (receptor del factor de crecimiento derivado de las plaquetas).6,7 La activación constitutiva de cualquiera de estos receptores de la tirosincinasa intervienen en la patogénesis y producción final del GIST.8 El principal criterio diagnóstico es el estudio IHQ CD 117 (95 % de positividad).2
Es un tumor frecuente entre los 55 y 65 años de edad, puede ser esporádico, familiar, o formar parte de un síndrome.1 Martin González y otros,2 en su publicación de cuatro casos de GIST de diferentes localizaciones (gástrica y rectal), tenían edades comprendidas entre 42 y 69 años. Las manifestaciones clínicas pueden variar dependiendo del sitio de origen del tumor, desde molestias hasta hemorragias gastrointestinales graves por erosión o infiltración de la mucosa. Se han descrito también fatiga y disfagia, así como metástasis en el hígado, omento, ganglios linfáticos, pulmón y cerebro, pero son poco frecuentes.2,5 Histológicamente el GIST, según el grado de mitosis y el diámetro tumoral mayor o menor de 5 cm y el índice de mitosis, permite evaluar criterios de malignidad.1,9
El tratamiento varía según el tamaño del tumor primario. De no poderse extirpar por riesgo de alta mortalidad, se puede usar terapia con imatinib como neoadyuvante para reducir el volumen del tumor, pero la toxicidad es elevada,2,9 por lo que la extirpación quirúrgica es lo indicado y posterior tratamiento. El imatinib por largo periodo es con el objetivo de inhibir la tirosincinasa. Si los márgenes quirúrgicos están libres de tumor, el peligro de recaída es mínimo a 5 años. Fletcher y otros1 han publicado un número importante de pacientes, que después de la resección intestinal por GIST primario, estuvieron libres de enfermedad a los 5 años en tumores resecados menores de 10 cm (54 %). El GIST esporádico se observa en adultos jóvenes, y se asocia con segundas neoplasias en el 13 % de los casos. Estas segundas neoplasias incluyen el carcinoma gastrointestinal, cáncer de próstata, de mama, renal y leucemias.1,6
El GIST se asocia a la neurofibromatosis tipo 1 (NF-1). También se asocia al síndrome de Carney-Stratakis, y la triada Carney: tumor estromal gastrointestinal epitelioide gástrico, paraganglioma extrasuprarrenal y condroma pulmonar. Los GIST asociados a neurofibromatosis tienden a ser multicéntricos, y son positivos al antígeno CD 117.10,11
Como tratamiento del GIST pediátrico se emplea el sunitinibe, inhibidores de la tirosina cinasa de segunda generación, aunque tienen alta toxicidad en edad pediátrica.12,13 Se describen insuficiencia cardiaca congestiva, hipertensión, náuseas, vómitos, neutropenia y alteración de la función tiroidea (hipotiroidismo subclínico), por lo que se emplean con cautela en Pediatría.14,15 Existen pocos casos publicados de GIST en niños, de ahí la importancia de nuestra publicación.
CONFLICTO DE INTERESES
Los autores declaran no tener conflicto de intereses en la realización del estudio.
REFERENCIAS BIBLIOGRÁFICAS
1. Fletcher CD, Berman JJ, Corless C, Gorstein F, Lasota J, Longley BJ, et al. Diagnosis of gastrointestinal stromal tumors: a consensus approach. Hum Pathol. 2002;33(5):459-65.
2. Martín-González MA, Fuentes Valdés E, Artiles Ivonet J, Solares María E, Martín-González AE, Lima M. Tumor del estroma gastrointestinal y cirugía. Rev Cubana Cir [serie en Internet]. 2008 Dic [citado 28 de Junio de 2016];47(3). Disponible en: http://scielo.sld.cu/scielo.php?script=sci_arttext&pid=S0034-74932008000300012&lng=es&nrm=iso&tlng=es
3. Shimomura M, Ikeda S, Takakura Y, Kawaguchi Y, Tokunaga M, Takeda H, et al. Gastrointestinal stromal tumors of the small intestine in pediatric populations: a case report and literature review. Pediatr Surg Int. 2010;26(6):649-54.
4. Joensuu H, Vehtari A, Riihimäki J. Risk of recurrence of gastrointestinal stromal tumor after surgery: an analysis of pooled population-based cohorts. Lancet Oncol. 2012;13(3):265-74.
5. Kindblom LG, Remotti HE, Aldenborg F. Gastrointestinal pacemaker cell tumor (GIPACT): gastrointestinal stromal tumors show phenotypic characteristics of the interstitial cells of Cajal. Am J Pathol. 1998;152(5):1259-69.
6. Corless CL, Heinrich MC. Molecular pathobiology of gastrointestinal stromal sarcomas. Annu Rev Pathol. 2008;3:557-86.
7. Miettinen M, Lasota J. gastrointestinal stromal tumors: review on morphology, molecular pathology, prognosis, and differential diagnosis. Arch Pathol Lab Med. 2006;130(10):1466-78.
8. Heinrich MC, Corless CL, Duensing A. PEDFRA activating mutations in gastrointestinal stromal tumors. Science. 2003;299(5607):708-10.
9. Joensuu H, Eriksson M, Sundby HK. One vs. three years of adjuvant imatinib for operable gastrointestinal stromal tumor: a randomized trial. JAMA. 2012;307(12):1265-72.
10. Gaal J, Stratakis CA, Carney JA. SDHB immunohistochemistry: a useful tool in the diagnosis of Carney-Stratakis and Carney triad gastrointestinal stromal tumors. Mod Pathol. 2011;24(1):147-51.
11. Gatta G, van der Zwan JM, Casali PG. Rare cancers are not so rare: the rare cancer burden in Europe. Eur J Cancer. 2011;47(17):2493-511.
12. Demetri GD. Gastrointestinal stromal tumor. In: De Vita VT Jr., Lawrence TS, Rosenberg SA. Cancer: Principles and Practice of Oncology. 9th ed. Philadelphia: Lippincott Williams & Wilkins; 2011. p. 1060-7.
13. Martel C, Forman D, Plummer M. Gastric cancer: Epidemiology and risk factors. Gastroenterol Clin North Am. 2013 Jun;42(2):219-40.
14. Mussi C, Ronellenfitsch U, Jakob J. Post-Imatinib surgery in advanced/metastatic GIST: is it worthwhile in all patients? Ann Oncol. 2010;21:403-8.
15. Demetri GD, Van Oosterom AT, Garret CR. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumors after failure of imatinib: a randomized controlled trial. Lancet. 2006;368(9544):1329-38.
Recibido: 8 de junio de 2016.
Aprobado: 21 de julio de 2016.
Caridad Verdecia Cañizares. Hospital Pediátrico Universitario "William Soler". San Francisco No. 10 112, Reparto Altahabana, municipio Boyeros. La Habana, Cuba.
Correo electrónico: caryverd@infomed.sld.cu

