










 Serviços customizados
Serviços customizados Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
La hipoplasia dérmica focal (HDF) o síndrome de Goltz fue descrita por primera vez en 1921 por Jessner; Lieberman en 1935 confirma el primer caso por medio de una biopsia. Años más tarde, en 1962, Goltz publica tres casos en mujeres con hipoplasia dérmica, herniación del tejido graso y defectos meso ectodérmicos. Finalmente, en 1963, Gorlin introdujo el término “hipoplasia dérmica focal” y describe los hallazgos histológicos que caracterizan al síndrome. Por esta razón, dicha genodermatosis también se conoce como síndrome de Goltz o síndrome de Goltz-Gorlin, pero puede confundirse con el síndrome del carcinoma basocelular névico, por lo que se prefiere el término de hipoplasia dérmica focal.1,2
En la década de los 60 del siglo xx, Goltz3 y Gorlin4 lo definieron en un síndrome caracterizado por atrofia e hiperpigmentación lineal de la piel, depósitos subcutáneos superficiales de tejido adiposo, lesiones papilomatosas múltiples en mucosas bucal, perianal y vulvar, así como en región perineal y piel periorificial, defectos en extremidades y anomalías de las uñas y le dieron el nombre de hipoplasia dérmica focal:
F/Female sex (sexo femenino),
O/Osteopathia striata (osteopatía estriada);
C/Coloboma (coloboma),
A/Absent ectodermis, mesodermis and neurodermis derived elements (ausencia de estructuras derivadas del ectodermo, mesodermo y neurodermo),
L/Lobster claw deformity (deformidad en pinza de langosta).3,4
Se asocian alteraciones óseas en el 80 % de los casos dada por deformidades en pinzas de langosta con sindactilia, hipoplasia o agenesia de los dedos, asimetría corporal, escoliosis y espina bífida, se presentan alteraciones dentarias como hipodoncia, defectos en la estructura del esmalte y retraso en la erupción dental. Suele haber retraso del crecimiento y delgadez.5
En el 95 % de los casos son esporádicos, sin embargo, se acepta una herencia autosómica dominante ligada al cromosoma X. Esto explicaría que el 88 % de los casos son mujeres. Para los varones homocigotos la enfermedad es letal y los casos registrados se deben a mutaciones nuevas en el gen PORCN (locus Xp11.23) o a alteraciones estructurales XXY, aunque 10 % de los casos afectados son hombres, debido a un mosaicismo pos-cigótico.1,2,3,4,5,6
La expresividad de la enfermedad es muy variable, puede encontrarse mínimas lesiones hasta compromiso generalizado en los miembros de una misma familia.(7,8,9,10,11,12.13,14,15)
El diagnóstico diferencial se realizó con: incontinencia pigmenti., síndrome Rothmund Thomson, nevo lipomatoso superficial, síndrome de Adams Oliver, aplasia cutis congénita. síndrome MIDAS (microftalmia, aplasia dérmica y esclerocórnea), síndrome EEC (displasia ectodérmica con ectrodactilia, labio y paladar hendidos). 1,15,16,17,18
El tratamiento es en equipo multidisciplinario.
PRESENTACIÓN DEL CASO
Paciente femenina de 4 años de edad, color de piel blanca, remitida de su área de salud para estudio y tratamiento a la consulta externa de cirugía maxilofacial del Hospital Pediátrico Docente Centro Habana, por presentar lesiones papilomatosas, en la región nasal y peribucal.
Examen físico positivo
Afecciones cráneo-faciales. Microcefalia, retraso mental, facies asimétrica, cabello escaso y quebradizo, orejas hipoplásicas con implantación baja, papilomatosis nasal y pericomisural con macrostomia izquierda.
Afecciones oculares: estrabismo y cataratas del ojo derecho con ceguera total del ojo izquierdo.
Alteraciones bucodentales. Microdoncia, oligodoncia, fusión dentaria e hipoplasia del esmalte. Amígdalas hipertróficas con múltiples lesiones papilomatosas (Fig. 1).
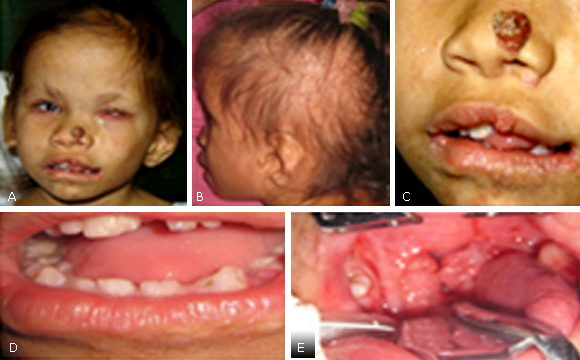
Fig. 1 - Microcefalia, facies asimétrica, estrabismo y cataratas del ojo derecho con ceguera total del ojo izquierdo. B. Cabello escaso y quebradizo, orejas hipoplásicas con implantación baja. C. Papilomatosis nasal y pericomisural con macrostomia izquierda. D. Microdoncia, oligodoncia, fusión dentaria e hipoplasia del esmalte. E. Amígdalas hipertróficas con múltiples lesiones papilomatosas.
Afecciones dermatológicas. Dermatosis diseminada, bilateral, simétrica. En extremidades superiores se disponen linealmente siguiendo toda la extremidad.
Región tóracoabdominal. Manchas hiperpigmentadas e hipocrómicas siguiendo las líneas de Blaschko.
Miembros inferiores. En la cara posterior del miembro inferior izquierdo presencia de masas abollonadas, rosadas, blandas, combinadas con lesiones acrómicas y atróficas, con la misma disposición lineal característica.
Región perianal. Se encontraron papilomas similares a los observados en la región naso-labial.
Lesiones esqueléticas. Sindactilia cutánea en la mano izquierda entre los dedos III y IV y sindactilia bilateral de pies; presencia de estrías longitudinales en los huesos largos y escoliosis tóracolumbar (Fig. 2).
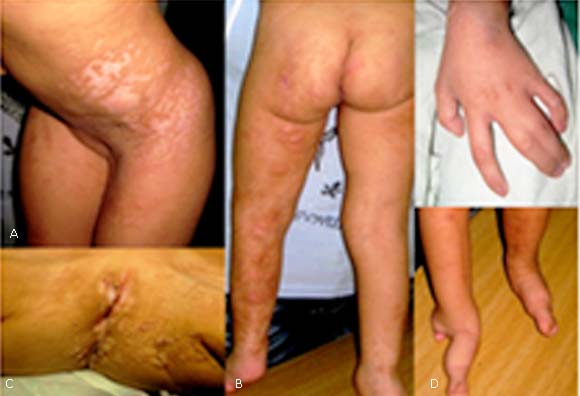
Fig. 2 - Manchas hiperpigmentadas e hipocrómicas siguiendo las líneas de Blaschko. B. Cara posterior del miembro inferior izquierdo presencia de masas abollonadas, rosadas, blandas, combinadas con lesiones acrómicas y atróficas. C. Región perianal (papilomas) D. Sindactilia.
Durante la anamnesis y examen físico encontramos importantes hallazgos que motivaron la valoración e interconsulta con otras especialidades: Genética, Dermatología, Oftalmología, Ortopedia, O.R.L, Estomatología y Neuro-pediatría. Se realizaron estudios que fueron concluyentes del síndrome de Goltz o hipoplasia dérmica focal. Entre los procedimientos quirúrgicos llevados a cabo por cirugía maxilofacial, dermatología y otorrinolaringología a la paciente se encontraron: la biopsia excisional de las lesiones de piel, las papilomatosas naso-labiales y amigdalotomías. (Fig. 3)
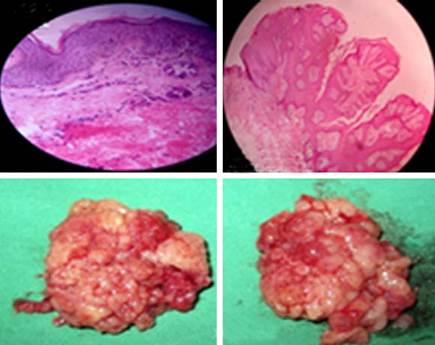
Las fotos presentadas estuvieron avaladas con el consentimiento de los padres.
DISCUSIÓN
El síndrome de Goltz se considera una genodermatosis muy rara, con menos de 300 casos publicados en la literatura científica. Por ello, no se conoce la prevalencia exacta a nivel mundial. Afecta predominantemente a las mujeres (9:1), y no se ha encontrado un predominio racial.1,2,3,4,5,6,7,15,16,17) En el Hospital Pediátrico Docente Centro Habana en 20 años de trabajo se han registrado solamente dos casos en el sexo femenino.
El 95 % de los casos son esporádicos y se presenta desde el nacimiento, sin embargo, se ha documentado la transmisión familiar con un modo de herencia dominante ligado al X por mutaciones en el gen PORCN (locus Xp11.23). Según Morales1) esto implica una mayor incidencia en mujeres (90 %) y letalidad en los homocigotos masculinos. Las pacientes del sexo femenino son heterocigotos o tienen un mosaicismo en la mutación del gen PORCN. En cambio, los varones (10-12 %) deben presentar forzosamente un mosaicismo somático debido a cualquiera de las siguientes: mutación de novo poscigótica, mutación de hemicromátide del cromosoma X o constitución cromosómica XXY.9,10,11,12,13,14,15,16,17,18
El diagnóstico es clínico, debido a que la topografía y morfología son características. En el caso de manifestaciones clínicas escasas, son de utilidad los hallazgos en la cavidad oral. Como pruebas adicionales pueden realizarse estudios moleculares, como el análisis de la secuencia genética en hombres y mujeres, y la hibridación fluorescente in situ o arreglos de hibridación genómica comparativa (CGH) en mujeres. El diagnóstico prenatal se aconseja en embarazos con riesgo elevado, es decir, cuando la mutación ya se ha identificado en la familia (ecografía). La biopsia de piel es útil para confirmar el diagnóstico. Se observa un pedículo fibrovascular, tejido conectivo laxo y numerosos vasos sanguíneos dilatados, rodeados de un infiltrado inflamatorio mixto. En la microscopia electrónica, en la dermis se observan numerosas estructuras lineares filamentosas, así como células grasas multiloculares, consideradas una forma de adipocitos inmaduros. Estudios ultraestructurales han demostrado un aumento de colágena tipo III en la dermis y ausencia de colágena tipo IV en la membrana basal. En la radiografía de huesos se hace necesario buscar osteopatía estriada.1,5,18,19,20) En el caso en estudio, las manifestaciones clínicas con los resultados de la biopsia por dermatología fueron concluyentes.
Las alteraciones cutáneas pueden tener una distribución bilateral lineal o reticular, que sigue las líneas de Blaschko, que corresponden a vías de migración neuronal durante el desarrollo embrionario. Las más características son cinco: distintos patrones de atrofia, lesiones estriadas, papilomas verrucosos tipo frambuesa, pólipos blandos y lipomas, que realmente son hernias de tejido adiposo por el adelgazamiento de la dermis. Estas áreas de herniación del tejido celular subcutáneo amarillento sobre la epidermis son características de esta entidad y se acompañan de atrofia cribiforme, rojiza o rojo-amarillenta con hiper- o hipopigmentación, telangiectasias, xerosis, foto sensibilidad y prurito.1,2
La lesiones se pueden ubicar en cualquier parte del cuerpo principalmente en piernas, antebrazos y mejillas (líneas radiadas desde los ángulos de la boca) localizados en áreas de mucosas y pliegues (perioral, perivulvar, perianal, periocular).Causan obstrucción de laringe, esófago o estómago.1,21,22,23,24,25
El diagnóstico radiológico de osteopatía estriada es clave en el diagnóstico en sujetos con expresión fenotípica mínima.
En cuanto al pronóstico, depende del grado de afección. Cuando esta es leve, tienen la misma esperanza de vida que la población general, según las alteraciones sistémicas asociadas. En cambio, cuando la afección es severa, los pacientes rara vez viven más allá de la infancia, y existen registros de abortos y mortinatos en la familia.1,17,20,21,22,23,24,25,26
Se concluye que el síndrome de Goltz o hipoplasia dérmica focal es una genodermatosis muy rara con una amplia variabilidad fenotípica debida muy probablemente a la inactivación aleatoria del cromosoma X, dependiendo del tipo de mutación presente en PORCN. Su manejo es multidisciplinario y el pronóstico depende del grado de afectación.17,23 En la actualidad se han publicado aproximadamente 200 casos del síndrome de Goltz en la literatura mundial.13,17,18,19,20,21,22,23,24,25,26,27

