Presentación de caso
Diagnóstico postmortem de un caso con síndrome hemofagocítico secundario
Postmortem diagnosis of a case with secondary hemophagocytic syndrome
Mercedes Cárdenas Bruno
1
*
, Myrna Moreno Miravalles
1
1Departamento de Anatomia Patológica. Hospital Pediátrico Docente “Juan Manuel Márquez”. La Habana, Cuba
RESUMEN
Introducción:
El síndrome hemofagocítico, llamado también linfohistiocitosis hemofagocítica o síndrome de activación macrofágica, es una grave enfermedad que se caracteriza por la activación exagerada del sistema inmune y aumento de la actividad linfocítica citotóxica y macrofágica, que puede ser potencialmente fatal.
Objetivo:
Describir un caso con este síndrome poco frecuente pero de alta mortalidad.
Presentación del caso: Paciente de 10 meses, nacido de parto eutócico, a término, normopeso, con antecedentes de ingreso a los dos meses por sepsis, con aumento de las transaminasas y adenopatías cervicales. Se realizó biopsia del ganglio cervical y se diagnosticó adenitis granulomatosa. En esta ocasión, cuatro días antes del ingreso comenzó con fiebre y decaimiento; al examen físico presentó tiraje intercostal bajo, polipnea superficial, hepatomegalia y esplenomegalia. Exámenes complementarios, presentó anemia, transaminasas, albúmina y proteínas totales elevadas; orina con pigmentos biliares y cuerpos cetónicos positivos; plaquetas 100 x 109. Los especialistas de gastroenterología plantearon una colestasis del lactante. El paciente falleció y en la necropsia se constató una hepatoesplenomegalia, hígado amarillento, adenopatías mesentéricas y peripancreáticas, pulmones hemorrágicos con aumento de consistencia; en el estudio microscópico se encontró en hígado, bazo, médula ósea, y ganglios linfáticos, histiocitos con hemofagocítosis.
Conclusiones:
El síndrome hemofagocítico es una enfermedad poco frecuente que muchas veces no se sospecha y pasa inadvertido, por lo tanto hay que pensar en dicha entidad porque tiene implicaciones pronósticas graves para el paciente, como puede ser un desenlace fatal.
Palabras-clave: síndrome hemofagocítico; hemofagocítosis; adenopatía
ABSTRACT
Introduction:
The hemophagocytic syndrome, also called hemophagocyticlymphohistiocytosis or macrophage activation´s syndrome is a serious disease characterized by exaggerated activation of the immune system and increased cytotoxic lymphocytic and macrophage activity, which can be potentially fatal.
Objective:
To describe a case with this rare syndrome with a high mortality rate, and the diagnosis was made postmortem in our hospital.
Case presentation:
A 10-month-old patient, born by natural delivery, with normal weight, with a history of admission at 2 months due to sepsis, with increased transaminases levels and cervical adenopathies. A cervical lymph node biopsy was performed, and granulomatous adenitis was diagnosed. On this occasion, four days before admission, he presented fever and weakness; physical examination revealed low intercostals retraction, superficial polypnea, hepatomegaly and splenomegaly. The complementary tests showed anemia, transaminases, albumin and total proteins with high levels; urine had bile pigments and positive ketone bodies; platelets 100 x 109. Gastroenterology specialists set out cholestasis of the infant. The patient died and at necropsy there were evidences of hepato splenomegaly, yellowish liver, mesenteric and peripancreatic adenopathies, hemorrhagic lungs with increased consistency. In the microscopic study, histiocytes with hemophagocytosis were found in liver, spleen, bone marrow, and lymph nodes.
Conclusions:
The hemophagocytic syndrome is a rare disease that often goes unnoticed. It has serious prognostic implications for the patient with a fatal outcome.
Key words: hemophagocytic syndrome; hemophagocytosis; adenopathy
INTRODUCCIÓN
La hemofagocitosis se define como la proliferación de las células histiocíticas que conservan su morfología normal, pero que despliegan una gran actividad fagocítica.(1
El síndrome hemofagocítico (SHF) inicialmente fue descrito en 1939 con el nombre de reticulosis medular histiocítica por Scott y Robb-Smith como una entidad clinicopatológica caracterizada por fiebre de inicio agudo, linfadenopatía generalizada, hepatoesplenomegalia y en estadios finales, ictericia, púrpura y pancitopenia, con proliferación sistémica de histiocitos con fagocitosis de eritrocitos.1,2 La etiología es desconocida pero se relaciona con anormalidad en la inmunorregulación que contribuye al descontrol y respuesta exagerada del sistema inmune.1,3,4,5 Se caracteriza por signos y síntomas de inflamación excesiva resultado de la disfunción de las células natural killer (NK) que lleva a sobreestimulación, proliferación y migración ectópica de células T.2) El síndrome hemofagocítico o linfohistiocitosis hemofagocítica (LHH), una entidad de difícil diagnóstico y con poco índice de sospecha, es una condición de hiperinflamación causada por una proliferación descontrolada de linfocitos e histiocitos activados por citocinas (interferón-gamma, factor de necrosis tumoral alfa, interleucinas (IL-1, IL-6, IL-10, IL-12, IL-18)( y factor estimulante de colonias de macrófagos), resultando en la activación de histiocitos con la subsecuente hemofagocitosis.4,6,7,8
El objetivo de este trabajo es describir un caso con este síndrome poco frecuente pero de alta mortalidad.
PRESENTACIÓN DEL CASO
Paciente de 10 meses, nacido de parto eutócico, a término, normopeso, con antecedentes de ingreso a los dos meses por sepsis, con aumento de las transaminasas y adenopatías cervicales. Se realizó biopsia del ganglio cervical y se diagnosticó adenitis granulomatosas. En esta ocasión, cuatro días antes del ingreso comenzó con fiebre y decaimiento; al examen físico se constató tiraje intercostal bajo, polipnea superficial, hepatomegalia de 3 cm y esplenomegalia. Exámenes complementarios: hemoglobina 7,0 g/L, transaminasa glutámica oxalacetica 805 UI/L, TGP 919 UI/L, albumina 22,2 g/L, proteínas totales 46,7 g/L, orina con pigmentos biliares y cuerpos cetónicos positivos, plaquetas 100 x 109 L. El ultrasonido abdominal informó hígado que rebasa el reborde costal 2 cm, aumento de la ecogenicidad y edema perivesicular. Los especialistas de gastroenterología plantearon una colestasis del lactante.
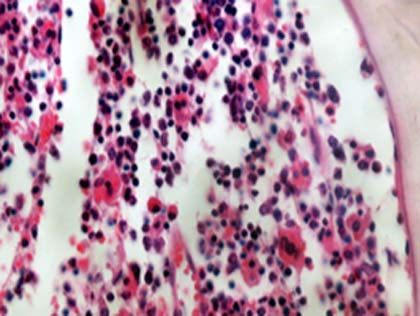
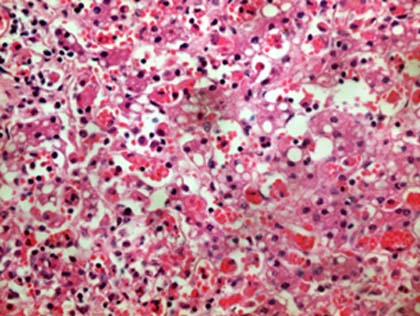
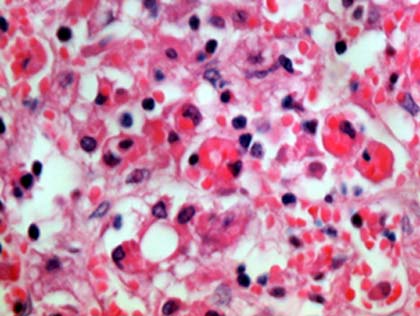
El paciente tuvo una evolución tórpida y fallece. En la necropsia se constató una hepatomegalia, esplenomegalia, hígado amarillento, bazo de consistencia aumentada, adenopatías mesentéricas y peripancreáticas, pulmones hemorrágicos con aumento de consistencia; en el estudio microscópico se encontró en hígado, bazo, médula ósea, y ganglios linfáticos histiocitos con hemofagocítosis (Figs.1,2,3) y escasos granulomas epitelioides en los pulmones. Se realizaron técnicas especiales en el ganglio linfático que ya habia sido biopsiado y en los pulmones para buscar microorganismos, todas fueron negativas.
DISCUSIÓN
El SHF es una grave enfermedad que se caracteriza por la activación exagerada del sistema inmune y aumento de la actividad linfocítica citotóxica y macrofágica, la que puede ser potencialmente fatal; existe una activación y proliferación descontrolada de células T, lo que causa una sobresecreción de citocinas de Th1, así como interferón gamma e IL-2 con activación de las células T y monocitos/macrófagos.1,9
El SHF es clasificado por muchos autores en primario y secundario, el primario se asocia a alteraciones genéticas específicas generalmente hereditarias o genéticas en la que la hemofagocitosis es la única manifestación y se asocia a un defecto central de la formación de perforinas y sintaxinas o a defectos como albinismo parcial y otros tipos de inmunodeficiencias y suele presentarse en los primeros meses de vida; el secundario, la forma adquirida que comprende cuadros secundarios hasta infecciones, neoplasias y enfermedades reumatológicas, se asocia a alguna enfermedad o condición subyacente y suele presentarse en la adultez.4,9).
Este síndrome se caracteriza principalmente por la presencia de pancitopenia, insuficiencia hepática, coagulopatía y diferentes síntomas neurológicos como somnolencia y crisis convulsivas, tiene una mortalidad significativa, la característica principal consiste en la acumulación de células tipo macrófagos bien diferenciados en diferentes órganos y tejidos como hígado, bazo, médula ósea y otros. El hallazgo citológico característico es la presencia de hemofagocitosis.10).
Para el diagnóstico se usan los criterios propuestos por el “Study Group of the Histiocyte Society”, citado por Peña7 y otros. Se revisaron en el 2004 y luego en el 2007.
Estos criterios son: fiebre, esplenomegalia, citopenia (> 2 líneas), Hb< 9 g/dL, plaquetas < 100 000 mm3, RAN< 1 000 mm3; hipertrigliceridemia ≥ 265 mg/dL, hipofibrinogenemia< 150 mg/dL, hemofagocitosis en médula ósea, ganglios linfáticos u otros, ferritina aumentada ≥ 500 μg/L, sCD25 elevado ≥ 2,400 U/mL, actividad NK disminuida o ausente. Deben estar presente al menos 5 de ellos.
El diagnóstico del caso que presentamos se hizo posmorten en el Hospital Pediátrico Docente “Juan Manuel Márquez”, La Habana, cuando se encontró en el estudio microscópico del hígado, bazo, médula ósea, y ganglios linfáticos, histiocitos con hemofagocitosis. El paciente no tenía antecedentes de enfermedad familiar o defecto genético conocido de esta enfermedad, pero por el antecedente de adenitis granulomatosa y la presencia de granulomas en los pulmones, planteamos que la hemofagocitosis en este caso es de tipo secundaria o adquirida.
Se concluye que el síndrome hemofagocítico es una enfermedad poco frecuente que muchas veces no se sospecha y pasa inadvertido por lo tanto hay que pensar en dicha entidad porque tiene implicaciones pronósticas para el paciente con un desenlace fatal.
REFERENCIAS BIBLIOGRÁFICAS
1.
Frenkel-Salamón M, Bolea-Murga V, Durán-Padilla MA. Síndrome hemofagocítico en pediatría. An Med Asoc Med Hosp ABC. 2001;46(3):137-41. Acceso: 07/07/2017. Disponible en: http://www.medigraphic.com/pdfs/abc/bc-2001/bc013g.pdf1.
[ Links ]
2.
Espinal D, Salinas F, Lanza L. Reporte de Caso: Linfohistiocitosis hemofagocítica Adquirida. Rev Scientifica. 2016;14(1):38-41. Acceso: 27/06/2017. Disponible en: http://www.revistasbolivianas.org.bo/pdf/rsscem/v14n1/v14n1_a10.pdf2.
[ Links ]
3.
Mostaza-Fernández JL, Guerra Laso J, Carriedo Ule D, Ruiz de Morales JMG. Linfohistiocitosis hemofagocítica asociada a infecciones virales: reto diagnóstico y dilema terapéutico. Rev Clín Esp. 2014;214(6):320-7. Acceso: 28/07/217. Disponible en: http://www.sciencedirect.com/science/article/pii/S00142565140012833.
[ Links ]
4.
Urías Estrella DM, González Pérez MC, Rascón Alcántar A, Díaz Reyes GA. Características clínicas del síndrome hemofagocítico en niños sonorenses. Bol Clin Hosp Infant Edo Son. 2016;33(2):49-53. Acceso: 07/07/2017. Disponible en: http://www.medigraphic.com/pdfs/bolclinhosinfson/bis-2016/bis162b.pdf4.
[ Links ]
5.
Hernández-Jiménez P, Díaz-Pedroche C, Laureiro J, Madrid O, Martín E, Lumbreras C. Linfohistiocitosis hemofagocítica: análisis de 18 casos. Med Clín. 2016;147(11):495-8. Acceso: 28/07/2017. Disponible en: http://www.sciencedirect.com/science/article/pii/S00257753163038405.
[ Links ]
6.
Izaguirre-González A, Sánchez-Sierra LE, Cerrato-Castro A, Flores-Irías J, Peña A. Síndrome Hemofagocítico Reactivo en Lactante Mayor. Reporte de Caso. Acceso: 27/06/2017 Disponible en: http://www.archivosdemedicina.com/medicina-de-familia/siacutendrome-hemofagociacutetico-reactivo-en-lactante-mayor-reporte-de-caso.php?aid=112676.
. [ Links ]
7.
Peña C, Valladares X, Cabrera ME. Síndrome hemofagocítico secundario: reporte de 5 casos. Rev Med Chile. 2013;141:1475-9. Acceso: 27/06/2017. Disponible en: http://www.scielo.cl/pdf/rmc/v141n11/art16.pdf7.
[ Links ]
8.
Vega J, Rodríguez MA, Goecke H, Santamarina M. Síndrome hemofagocítico en un trasplantado renal con síndrome de Alport. Rev Med Chile. 2013;141(4):519-24. Acceso: 28/07/2017. Disponible en: http://www.sciencedirect.com/science/article/pii/S1699258X130020528.
[ Links ]
9.
Beffermann N, Pilcante J, Ocqueteau M, Sarmiento M. Síndrome hemofagocítico adquirido: reporte de casos de cuatro pacientes adultos tratados con protocolo HLH 94-04 y revisión de la literatura. Rev Med Chile. 2015;143:1172-8. Acceso: 07/07/2017 Disponible en:http://www.scielo.cl/pdf/rmc/v143n9/art10.pdf9.
[ Links ]
10.
Alonso Castillo A. Síndrome hemofagocítico en pediatría. La Habana: Hospital "William Soler", Catedra de Pediatría; 2010. Acceso: 07/07/2018 Disponible en: http://www.sld.cu/galerias/pdf/sitios/williamsoler/sindrome_hemofagocitico_en_pediatria__(revision).pdf10.
[ Links ]











 Serviços customizados
Serviços customizados Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
Permalink